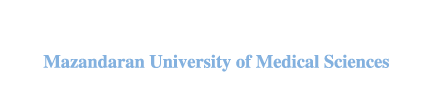Volume 8, Issue 4 (2022)
Pharm Biomed Res 2022, 8(4): 237-258 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Kehinde O A, Damilare B I, Ogunlana A, Mojeed Ayoola A, Opeyemi Emmanuel A, Temitope Isaac A. Inhibitors of α-glucosidase and Angiotensin-converting Enzyme in the Treatment of Type 2 Diabetes and its Complications: A Review on in Silico Approach. Pharm Biomed Res 2022; 8 (4) :237-258
URL: http://pbr.mazums.ac.ir/article-1-453-en.html
URL: http://pbr.mazums.ac.ir/article-1-453-en.html
Oyedele Abdul-Quddus Kehinde1 

 , Boyenle Ibrahim Damilare1 , AbdeenTunde Ogunlana1 , Ashiru Mojeed Ayoola2 , Atanda Opeyemi Emmanuel1 , Adelusi Temitope Isaac * 1
, Boyenle Ibrahim Damilare1 , AbdeenTunde Ogunlana1 , Ashiru Mojeed Ayoola2 , Atanda Opeyemi Emmanuel1 , Adelusi Temitope Isaac * 1
, Boyenle Ibrahim Damilare1 , AbdeenTunde Ogunlana1 , Ashiru Mojeed Ayoola2 , Atanda Opeyemi Emmanuel1 , Adelusi Temitope Isaac * 1
1- Department of Biochemistry, Faculty of Basic Medical Sciences, Ladoke Akintola University of Technology, Ogbomoso, Nigeria.
2- Department of Chemical Sciences, College of Natural and Applied Science, Fountain University, Osogbo, Nigeria.
2- Department of Chemical Sciences, College of Natural and Applied Science, Fountain University, Osogbo, Nigeria.
Keywords: α-glucosidase, Angiotensin-converting enzyme, In silico drug discovery, Pharmacological activity, ADMET
Full-Text [PDF 1677 kb]
(950 Downloads)
| Abstract (HTML) (1638 Views)
Full-Text: (925 Views)
Introduction
The aim of this review was to elucidate the pathophysiological relationship between type 2 DM (T2D) and hypertension and then summarize several studies conducted on in silico screening of α-glucosidase inhibitors in recent years with the objective of recommending them for further preclinical and clinical studies. We further explained the influence of α-glucosidase competitive and allosteric inhibitors on the treatment/management of diabetes and its downstream complications. In addition, since diabetic patients have been found to suffer from chronic complications, such as hypertension and diabetic nephropathy, we also investigated recent computational studies on angiotensin-converting enzyme (ACE) since inhibitors of this enzyme could synergistically prevent hypertension and progression of diabetic nephropathy. Moreover, we noticed that most authors failed to perform in silico ADMET studies of the hit compounds, which is essential in the early phase determination of the safety profile of drug candidateFs and filtering out leads with no favorable pharmacokinetics before they could be subjected to further preclinical ADMET studies. Hence, we carried out in silico meta-analysis on the ADMET profile of the reported compounds and recommend them for further preclinical trials.
The α-glucosidase enzyme has been identified as an efficient molecular target in treating T2D. T2D, also referred to as non-insulin-dependent diabetes, occurs in more than 90% of diabetic patients, and about 80% of them are usually obese [1]. Non-insulin-dependent diabetes mellitus poses a significant threat to the health of humans as a result of its higher rate of occurrence, chronic implication, and complications [2]. Statistical data from the World Health Organization (WHO) highlighted the prevalence of T2DType. In 1980, statistics revealed that over 4.7% of the world population had T2D, and a progressive rise occurred in 2014 after a study showed the percentage had increased to about 8.5% of the world population [3].
Also, α-glucosidase is a key enzyme in carbohydrate digestion responsible for the breakdown of maltose with the concomitant release of glucose [4]. This digestive enzyme acts on the α-1,4 glycosidic bond of disaccharide, a different enzyme from β-glucosidase [5]. Hence, identifying and targeting this digestive enzyme for inhibition to reduce the rate, at which glucose is absorbed in the intestine could be an important therapeutic measure in managing T2D [6].
Several clinically approved drugs against α-glucosidase, such as acarbose, miglitol, and voglibose have been used for the treatment of T2D [4]. These drugs have been designed to act as antidiabetic agents inhibiting the digestion of carbohydrates and slowing down the absorption of glucose. Consequently, this allows the blood glucose level to be maintained constant. Although some clinically approved drugs have been reported against α-glucosidase, these standard drugs are associated with undesirable side effects. Sheliya et al. in 2015 reported that the use of acarbose-a clinically and the widely used antidiabetic drug are associated with several side effects, such as abdominal swelling, pneumatosis cystoides, diarrhea, and flatulence [7]. Many studies have been conducted recently to identify new drug candidates from plant extracts as an alternative to the existing standard drugs with α-glucosidase inhibitory activity [8]. Pharmacological agents derived from plant extracts are increasingly gaining acceptance in the management of diabetes because they are easily accessible and do not require laborious pharmaceutical synthesis compared to their synthetic counterparts [9].
Also, it has been reported that more than 75% of patients with diabetes suffer from microvascular complications, such as hypertension [10], and need antihypertensive drugs, such as ACE inhibitors (ACEI) to treat their condition. Moreover, aside from the antihypertensive effect of this drug, they also help delay the progression of nephropathy in diabetic patients who are normotensive and hypertensive. In addition, 25-40% with type 1 or 2 diabetes have diabetic nephropathy after 20-25 years of the onset of the disease [11]. The distinguishing clinical characteristics of diabetic nephropathy involve a gradual increase in the level of albuminuria and a declined glomerular filtration rate (GFR), which occur with an accompanying increase in blood pressure and eventually cause end-stage renal failure [12]. Angiotensin-2, a potent vasoconstrictor produced as a result of ACE activity, causes diabetic nephropathy by inducing hypertrophy in mesangial and epithelial cells [13] and also promoting the production of transforming growth factor (TGF-β), which has been identified as a causative agent for glomerulosclerosis [14]. ACEIs are the most widely used agents in diabetic patients due to their capacity to slow the deterioration of renal function and decrease proteinuria [15]. In a comprehensive review by some group of scientists, findings support the use of ACE inhibitors in patients with diabetes mellitus and hypertension because aside from its antihypertensive property, it also prevents chronic complications, such as diabetic nephropathy as a result of its renoprotective effect [16].
Pathophysiological interplay between T2D, hypertension, and diabetic nephropathy
Diabetes, most especially T2D represents a global health problem of epidemic proportions and arises as a result of insulin resistance [17]. Enzymes responsible for the digestion of carbohydrates have been recognized as an important target in the management of T2D. After the ingestion of carbohydrates, the digestive process of polysaccharides begins with a breakdown into oligosaccharides by salivary α-amylase and subsequently degraded into disaccharides with pancreatic α-amylase. The α-amylase enzyme hydrolyzes the α-bonds of large α-linked carbohydrates and is found mostly in saliva and pancreatic juice [18]. Disaccharides in the small intestine are catabolized to monosaccharides, such as glucose, fructose, etc. by α-glucosidase produced in the intestinal epithelial cells. Hence, the key enzyme responsible for the final breakdown of polysaccharides to glucose is α-glucosidase. Studies have shown that inhibitors of α-glucosidase could retard the uptake of glucose, decrease postprandial hyperglycemia, and consequently be useful in the management of T2D [19, 20].
One of the major vascular complications of T2D is hypertension caused by high blood pressure. Also, 70% of diabetic patients also develop high blood pressure [10]. It has been suggested that high blood glucose levels in the bloodstream could enhance the nonenzymatic glycosylation of several molecules and stimulate the production of reactive oxygen species (ROS), which may overwhelm the body’s antioxidant system and subsequent manifestation of diabetic chronic complications [21]. Recently, Marc et al. reported that blindness, renal disease, and cardiovascular disease are the chronic complication of diabetes [22]. Among the risk factors of cardiovascular disease is hypertension, which is approximately twice as frequent in diabetic patients than in those without the disease [23]. Hence, regulating postprandial hyperglycemia is an important therapeutic strategy in the management or treatment of diabetes and synergistically could enhance a holistic decrease in high blood pressure related to diabetes as shown in Figure 1 [24].

Also, the administrations of pharmacological agents inhibiting the renin-angiotensin-aldosterone system (RAAS) have been reported to hinder the onset of T2D in high-risk populations [25, 26]. In the renin angiotensin aldosterone system, the hepatic angiotensinogen is cleaved by renal synthesized renin to produce Ang I, which is then converted to a strong vasoconstrictor-Ang II in the lung by the action of ACE and thereby causing high blood pressure. Although the use of a pharmacological intervention that causes blockage of the RAAS pathway to reduce high blood pressure in subjects with hypertension, some reports obtained from rodents and cell experiments suggest that ameliorating insulin sensitivity by improving the function of adipose tissue and enhancing β-cell function through improved islet perfusion and anti-fibrotic effect are the proposed mechanism of action, by which therapeutic agents blocking RAAS pathway could reduce the risk of T2D [27-30]. The administration of angiotensin-converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) blocking the RAAS system reduced the risk of T2D in hypertensive patients [31-33].
T2D and hypertension caused by hyperglycemia and high blood pressure respectively have also been linked to diabetic nephropathy (DN). High blood glucose could lead to the formation of Amadori products (altered proteins) and advanced glycation end products, which are part of the component of DN. Furthermore, activation of the electron transport chain caused by hyperglycemia can lead to increased generation of ROS, which may be the initiating agent in the development of diabetic complications. The hemodynamic pathway involving the formation of growth factors, hypertrophy, extracellular matrix accumulation, cytokines, proteinuria, and ROS has been implicated in the progression of DN [34]. Hyperglycemia, advanced glycation end products (AGE), and ROS could work in harmony to cause the release of cytokines and growth factors through a signal transduction pathway that involves protein kinase C and nuclear transcription factor (NF-KB). Transforming growth factor (TGF-β) could induce hypertrophy of the kidney cells and accumulation of extracellular matrix proteins [35]. Similarly, angiotensin II could induce hypertrophy in mesangial and epithelial cells [13], and enhance the formation of TGF-β, which are part of the key players in DN [14]. In addition, studies have shown that the strong vasoconstrictor Ang II produced as a result of the activation of the renin-angiotensin aldosterone system is involved in almost all stages of the development of DN [36]. The emergence of hypertension in a diabetic patient could increase the risk of developing end-stage renal disease five to six folds compared to patients who are hypertensive alone [37]. Inhibitors of the renin-angiotensin system, such as ACEI and angiotensin receptor blockers (ARB) have been shown to impede progressive kidney impairment and prevent the onset of end-stage kidney disease in patients with DN [38].
Competitive inhibitors of α-glucosidase
The experimental x-ray crystallography technique has revealed the architectural structure of α-glucosidase, which consists of N- and C-terminal catalytic domains [39, 40]. Researchers have thus identified the active residues in the catalytic domain of this enzyme and labeled them as a therapeutic target for the design of antidiabetic agents in the drug discovery process. The vast majority of reports on the screening of α-glucosidase inhibitors that use the molecular modeling approach are focused on the recognition of small molecules derived from natural plant extracts acting as competitive inhibitors or blockers of the enzyme’s active site.
A good example of a competitive inhibitor of α-glucosidase was reflected in the work where Epicatechin-3,5-O-gallate (ECDG) derived from Orostachys japonicus binds to the receptor active site [8]. A number of compounds derived from Orostachys japonicus were docked against the α-glucosidase binding site; however, ECDG proved to be the best inhibitor of the enzyme with autodock score of -10.17 kcal/mol. The major interaction contributing to this high binding affinity was nine hydrogen bonds with six amino acids, including Pro224, Gln233, Asn236, His240, Asn241, and Pro304. The molecular docking result suggests that ECDG may inhibit the catalytic reaction of α-glucosidase by covering the active site pocket. Molecular dynamics of 10 ns were also executed to uncover the behavior and dynamics of this receptor-ligand binding process. The molecular dynamic result showed that ECDG mainly had 1-3 hydrogen bonds, with receptor residues in the stable complex. Asn236 was found to be a critical residue as it participated in the formation of hydrogen bonds throughout the simulation. The root means square deviation (RMSD) was evaluated as 0.30 nm after 10 ns, which represents the stability of the complex. This result revealed that ECDG may be a potential drug candidate for α-glucosidase, which could be used in the treatment of T2D.
Xu et al. [41] demonstrated the inactivation of α-glucosidase with Hydroxysaflor yellow A (HSYA) using a molecular dynamics study. The targeted receptor and HSYA extracted from Carthamus tintorius were used in the docking experiment to obtain the best ligand pose. The result was then used to conduct a molecular dynamic simulation of 10 ns, which indicated the active site interaction of HSYA with G217, A278, H279, and G280. The molecular modeling result was shown to be consistent with their enzyme kinetics study, which proves that HSYA might interrupt the substrate binding activity of α-glucosidase in a competitive manner and hence acts as a potential anti-diabetic agent in the treatment of T2D.
Xiong et al. [42] assessed the inhibitory effect of α-ketoglutaric acid (AKG) on α-glucosidase. The binding pocket of α-glucosidase was assessed for AKG docking with α-glucosidase-like-proteins, using the sequence alignment program in the HHblits server. VMD was used to determine the position of the bound ligands in the enzyme pocket. The overlapping regions around the ligand-binding points and the α-glucosidase pockets were identified to define the AKG binding site. The molecular docking process was carried out using AutoDock Vina version 1.1.2 and the result showed that AKG was stably positioned at the existing pocket of the enzyme. They also performed a molecular dynamic simulation of 10 ns, which reveals that TYR313, ASN412, and ARG439 formed hydrogen bonds with AKG and thereby facilitating its binding and stability. They validated this molecular modeling result with an experimental enzyme kinetic study, which shows that AKG competitively inhibits α-glucosidase.
In 2019, Choudhary DK, et al. [43] examined the inhibitory activity and mechanism of polyphenols extracted from fava bean on α-glucosidase. They performed molecular docking on all the extracted polyphenols and discovered that only two compounds (gallic acid and catechin) had the highest binding affinity (-6.58 and -7.25 kcal/mol, respectively) in comparison with the standard acarbose drug (-6.35 kcal/mol). Furthermore, both compounds were found to have four hydrogen bonds and the critical amino acid interaction responsible for their binding affinity was located at the active site of the enzyme with Asn324, Asn326, and Arg197 for gallic acid and Asp198, Asn324, Asp326, and Tyr63 for catechin. Molecular dynamics with GROMACS was used to run a simulation that spanned 50 ns and the result substantiated the interaction between gal:α-glucosidase and cat:α-glucosidase as stable complexes with the RMSD varying between 0.15 and 0.35 nm throughout the simulation period. These findings revealed that ethanolic seed extract from fava beans could be an efficient α-glucosidase inhibitor.
Gong et al. [4] evaluated the inhibitory effect of hesperetin (a flavonoid derivative) on α-glucosidase based on computational molecular docking and dynamic simulation. The docking experiment showed that hesperetin was tightly bounded and buried into the active pocket of the enzyme with a binding affinity of -7.1 kcal/mol. The docked structure was used to run 10 ns MD simulations to estimate how tightly the ligand binds to α-glucosidase. The MD simulation revealed the critical amino acids responsible for hesperetin: α-glucosidase binding, which were Lys155, Ser156, Phe157, Gly159, Phe231, Gly159, Phe231, Asn241, Ala 278, His279, Glu304, Thr307, Ser308, Pro309, Phe311, and Arg312. After the MD simulation, the distance profile analysis was carried out to determine whether the α-glucosidase pocket was blocked by hesperetin. Interestingly, the distance profile revealed that five groups of hesperetin were found in close contact (short distance below 6Å) with six crucial residues (Lys155, Asn241, Glu304, Pro309, Phe311, and Arg312) in the active site ofα-glucosidase. This analysis showed that hesperetin blocked the binding pocket of the receptor, thereby inhibiting its catalytic activity. The in silico result was also found to be in agreement with their experimental enzyme kinetic studies, which uncovered the binding and inhibitory mechanism of hesperetin as an α-glucosidase competitive inhibitor.
In another study, Rahman et al. [5] investigated the inhibitory potential of 32 isolated alkaloids from plants against α-glucosidase using a molecular docking screening process. The criteria for their selection of these compounds were based on already reported in vitro and in vivo activities of these alkaloids on α-glucosidase inhibition. The docking process was done by the molecular operating environment (MOE) software for the prediction of the binding affinities of the selected 32 alkaloids against α-glucosidase. The results revealed nummularine-R and vindoline as the most potent compounds with docking scores of -14.5691 and -13.2250, respectively. These docking scores were also found to be very close to standard α-glucosidase inhibitors-acarbose and miglitol (-14.7983 and -15.4423, respectively). Furthermore, both ligands recorded good interactions with the active site of the target enzyme as the most potent compound (nummularine-R) formed four hydrogen interactions with Gln121, Met122, Arg331, and Gly546 while the second most potent compound (vindoline) had three hydrogen bonds and one arene-arene interaction with amino acids Ala93, Ala97, Gln121, and Trp126. The results of these docking screenings were consistent with previously reported in vitro antidiabetic experiments [44, 45], which revealed the promising potential of nummularine-R and vindoline as α-glucosidase inhibitors.
In another study, Laoud et al. identified some novel inhibitors of nt-MGAM using QSAR, docking, and ADME for their virtual screening process. Firstly, they performed molecular docking of a set of salacinol derivatives into the active pockets of N-terminal α-glucosidase, and then the authors used an atom-based QSAR model to identify pharmacophoric features of salacinol derivatives responsible for the enzyme inhibition. Virtual screening was subsequently carried out to screen potential compounds with similar chemical scaffolds in the ZINC database. Eventually, ten hit compounds (ZINC 39979739, ZINC 72426723, ZINC 39976379, ZINC 02881852, ZINC 91462504, ZINC 65528728, ZINC 63884379, ZINC 65371910, ZINC 72473431, and ZINC 39371167) were identified and satisfactory ADME profiles were then obtained for this drug-like compounds, which indicated that the salacinol analogues may be a promising drug in the treatment of T2D [46].
In other research, Zafar et al. examined the α-glucosidase inhibitory potential of some isolated alkaloids. MOE software was used to obtain the tertiary structure of the biological target and molecular docking was used to dock the 37 isolated alkaloids to the modeled protein. The result revealed that compounds 17 (oriciacridone F) and 24 (o-methyl mahanine) possess striking interactions with the active amino acid of the enzyme and the interaction demonstrated were similar to those of the standard drugs (acarbose and miglitol), making them potential lead compound against α-glucosidase [47].
There are also theoretical reports on the use of molecular modeling experiments for the assessment of the inhibitory potential of peptides against α-glucosidase. An example of this could be found in the study by Mollica et al. [48], where they assessed the screening of a combinatorial peptide library that could act as a promising inhibitor of α-glucosidase through binding to the enzyme’s active pocket. Hence, in order to identify novel peptide candidates that could act as potent inhibitors of the molecular target, the top 12 hit compounds were selected for MD simulations from the docking result based on their higher binding affinity. They performed an MD simulation of 20 ns to analyze the stability of the peptides with α-glucosidase. After the simulation, four peptides (H-Phe-Arg-Asn-NH2, H-Trp-Arg-Ala-NH2, H-Lys-Arg-Met-NH2, and H-Asn-Arg-Asn-NH2) were chosen as the final hit compounds on the basis of their lower RMSD value, which signals higher stability compared to other peptides that passed through the MD simulation. Interestingly, the in silico result was also found to be consistent with their in vitro experiment with the four peptides having inhibitory potency that is very similar in relation to standard acarbose. This study showed that peptides could act as potential agents in the treatment of diabetes.
Allosteric inhibitors of α-glucosidase
Targeting the allosteric site of enzymes or proteins may emerge as a compelling alternative inhibition mechanism of some ligands other than binding to the receptor active site and some examples of these inhibitors have been reported in the modulation of α-glucosidase.
Recently, in 2019, Wan et al. [49] investigated the inhibitory potency of phloroglucinol against α-glucosidase using enzyme kinetics and molecular dynamics study. Earlier in their experiment, enzyme kinetic results had shown that phloroglucinol is a reversible inhibitor of α-glucosidase in a dose-dependent but non-competitive mode. Thus, in order to assess this mode of inhibition in silico, they performed molecular docking and MD simulation study on α-glucosidase. The tertiary structure of the enzyme (Saccharomyces cerevisae) was obtained using homology modeling as the x-ray crystallographic structure was unavailable at the time. Based on the predicted phloroglucinol-binding residues (Tyr71, His111, Phe177, Arg212, Asp214, Thr215, Gly217, Leu218, Glu276, His348, and Asp349), AutoDock Vina v.1.1.2 was used for the molecular docking of phloroglucinol to α-glucosidase to obtain the best pose of the protein-ligand complex. The binding affinity of the complex was calculated as -7.48 kcal/mol and the best pose was subjected to MD simulation of 10 ns. The docking and MD simulations revealed that phloroglucinol was almost completely localized in the α-glucosidase binding site while interacting with a single binding site near the active pocket. Furthermore, the phloroglucinol binding was found to be more stable at the entry point of the active site (rather than inside the enzyme’s active pocket) with the distant profile analysis revealing the distance between the interaction of residues and relative bound inhibitor to be less than 6Å. They concluded that phloroglucinol is a promising allosteric inhibitor of α-glucosidase, which could interfere in the normal catalysis of the enzyme and therefore, could be used in the treatment of T2D.
In another study, Ding et al. [50] examined the synergistic inhibition mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Prior to their in silico assessment of the two compounds on α-glucosidase, they carried out an enzyme kinetic inhibition study, which revealed that oleanolic acid and ursolic acid exhibited effective inhibitory activities with IC50 estimated as (6.35±0.02)×10-6 and (1.69±0.03)×10-5 molL-1, respectively in a reversible and non-competitive manner. This result was further validated with their docking experiment, which showed that the two compounds bind to different allosteric sites on α-glucosidase in cavity 2 and cavity 4, respectively. Oleanolic acid interacted with allosteric residues, such as Trp14, Lys12, Ser295, Ala289, His258, Tyr292, Lys262, Val265, Ile271, and Glu270 with -4.63kcal/mol while ursolic acid bound with amino acids Trp465, Glu405, Val407, Lys410, Asn411, Ser179, Arg180, Gln67, Gln66, and Met69 with -4.37 kcal/mol. This protein-ligand interaction does not correspond to the amino acid residues on the active pocket of the enzyme, which were positioned in cavity 1 and thereby allowing allosteric binding to perturb the conformational dynamics of the enzyme leading to a reduction in the catalytic activity of α-glucosidase. The in silico result coupled with in vitro study revealed the inhibitory mechanism of oleanolic acid and ursolic acid on α-glucosidase and their management in the treatment of T2D.
Meena et al. [51] assessed the α-glucosidase inhibitory potential of a synthetic flavone derivative 2-(benzo [d] [1, 3] dioxol-5-yl)-4H-chromen-4-one (BDC) with their in vitro enzyme kinetic and in silico studies. The mode of the mechanism of BDC had been earlier revealed by authors in their in vitro experiment as non-competitive with the maximum α-glucosidase inhibitory potential of the ligand 22.4 folds over the maximum competitive inhibition depicted by standard acarbose drug (27.6 to 669.57 μM). In other to substantiate and show the allosteric interaction of BDC with α-glucosidase, they performed in silico molecular docking experiment. The active sites of the enzyme were determined using CASTp 3.0 web server.
The docking experiment of acarbose with the receptor revealed that acarbose competes for the active pocket of the α-glucosidase enzyme with a minimum binding affinity of -9.23 kcal/mol. Two hydrogen bonds (LYS156:H23 (2.03Ao) and SER241:HN (2.02Ao)) were recognized as the interaction between the receptor and the ligand while interactions are electrostatic, Van der Waals, and hydrophobic interactions. Contrary to this mode of interaction in acarbose, BDC depicted allosteric binding with its interaction on α-glucosidase found to be different from the active pocket residues. Two hydrogen bonds were also observed between LYS373:H22 (2.077Ao) and LYS568:H23 (2.09Ao) of the protein-ligand complex while other interactions are electrostatic/hydrophobic bonds. The binding of BDC [-8.64kcal/mol) showed that the potential drug candidate inhibitory effect was significant compared to the standard clinically approved acarbose (-9.23 kcal/mol). Furthermore, Lipinski’s rule of five was used to evaluate the drug-likeness profile of the ligand and by obeying all rules coupled with its high binding affinity with α-glucosidase, the authors concluded that BDC could be a promising lead compound in the management of diabetes.
Angiotensin-converting enzyme (ACE) structural information, inhibitors, and molecular modeling
Investigation of the molecular structure of ACE has revealed that the human angiotensin-converting enzyme comprises two isoforms: somatic ACE, which consists of two catalytic homologous domains (N- and C-terminal with 60% similarities) [52], and testis ACE, which has only one domain that is structurally similar to the C-terminal catalytic domain of somatic ACE [53]. The two homologous domains of ACE could hydrolyze angiotensin-1; however, both have different pharmacological potentials as they have different substrate specificities. Fuchs et al. revealed that C-domain is enough to regulate blood pressure in vivo [54] and is thus a major site for angiotensin-2 production. Conversely, the N-domain is more specific for the control of haemopoietic stem cell proliferation and differentiation due to its breakdown of anti-fibrotic haemoregulatory peptide AcSDKP [55, 56]. Hence, it is important that the C-domain of ACE be given more preference in the selection of a target for ACE inhibitors during the rational drug discovery process.
Computational molecular modeling technique has been used in the screening of ACE inhibitors. Most of the investigations on the computational screening of ACE inhibitors have focused on the recognition of peptides gotten from hydrolysates of protein or natural sources. For example, Yu et al. [57] explored the ACE-inhibitory potential of some selected tripeptides obtained from in silico gastrointestinal proteolytic cleavage of Larimichthys crocea nebulin protein. The 168 unreported tripeptides derived from the proteolytic process were subjected to drug-likeness and ADME screening. Eventually, four tripeptides (EGF, MDY, GDL, and HGR) were chosen as they have reasonable molecular weight and better pharmacokinetic profiles, such as human intestinal absorption, water solubility, and CYP450 inhibition compared to others. Hence, these four peptides were selected for molecular docking experiments. The crystal structure of ACE in complex with lisinopril (1o86) was retrieved from the protein data bank and the authors performed the docking protocols with CDOCKER PROGRAM. The binding affinity of -92.4782, -90.8416, -80.8434, and -83.4035 kcal/mol was obtained for HGR, EGF, GDL, and MDY tripeptides, respectively. On the basis of binding affinity (-92.4782 kcal/mol) and the number of intermolecular hydrogen bonds (14 H-bond), Yu et al. selected HGR as the best ACE inhibitor and subjected it to further in vitro experiments.
In another study, De Oliveira et al. demonstrated the ACE-inhibitory mechanism of six peptides (ALNEINQFYQK, NAVPITPTLNR, FALPQYLK, FFVAPFPEVFGK, YLGYEQLLR, and HQGLPQEVLNENLLR) derived from hydrolysis of milk caseins. The crystal structure of human ACE (pdb id:1o86) was obtained from the RCSB protein data bank and used as a molecular target for the docking process. De Oliveira et al. performed a docking experiment of the peptide ligands to the active site of the N- and C-catalytic domains of ACE using AutodockVina. They reported that out of the entire 12 docked complexes, only peptide FALPQYLK N-oriented pose failed to bind with ACE while others recorded good interaction with the enzyme active site. In order to examine the dynamics of these complexes, they performed a molecular dynamic simulation of 50 ns using GROMACS version 5.1.4. According to RMSD analysis (less than 3Å), no noticeable conformational change of all the complexes was observed throughout the simulation with respect to their initial docked pose. The RMSF analysis of their in silico study showed that some amino acids located around the ACE extremities have high fluctuation regardless of the ligand geometry or conformation and do not inhibit ACE. They then proposed that this finding could give insight into a computational prediction of promising and non-promising ACE inhibitory peptides obtained from other sources [58].
In another research, Tahir et al. [59] assessed the ACE inhibitory potential of some peptides derived from a blood pressure-regulating honey protein (MRJPI). I-TASSER homology modeling server was used to determine the tertiary structure of MRJPI. However, in order to improve the quality of the modeled structure, optimization of the receptor was carried out with molecular dynamics of 30ns. The RMSD, RMSF, and B-factor analysis revealed the structural stability of MRJPI and was considered to be suitable for further in silico assessment. To identify potential inhibitory peptides, an AHTpin server based on a scoring vector machine of regression models was used for the proteolysis of the optimized protein. They then performed a docking study of all derived peptides to evaluate their binding affinity with ACE. Tahir et al. then identified the hit ligand “EALPHVPIFDR” that had the highest ACE-inhibitory potential from its binding affinity value (-58.29 kcal/mol). Interestingly, from the knowledge of the peptides-ACE interaction, the researchers discovered that Tyr residue was particularly a crucial residue, which could be a therapeutic target during ACE-inhibition rational drug design process.
In another study, Auwal et al. [60] examined the ACE inhibitory potential of five peptides (Ala-Leu-Gly-Pro-Gln-Phe-Tyr, Lys-Val-Pro-Pro-Lys-Ala, Leu-Ala-Pro-Pro-Thr-Met, and Asp-Val-Leu-Ile-Gln) derived from stone fish protein hydrolysate. Their in vitro studies had shown that the five peptides are promising ACE inhibitors with satisfactory IC50 values. Hence, in order to substantiate this finding, the authors performed a molecular docking experiment of the potential drug candidates with Glide XP software. Interestingly, Auwal et al. docking result was found to be consistent with their in vitro experiment with peptide sequence Ala-Leu-Gly-Pro-Gln-Phe-Tyr showing the highest binding affinity (-10.374 kcal/mol) and lowest IC50 value. Likewise, the Gly-His-Pro-Val-Leu sequence had the lowest inhibitory effect both in silico (-8.635 kcal/mol) and in vitro levels. In this study, the scientists demonstrated the effectiveness and promising inhibitory activities of a peptide derived from stone fish. In another research, a group of researchers derived 126 potential ACE-inhibitory peptides from Tobarodespacifus using in silico and bioinformatic tools. After their pharmacokinetic and pharmacodynamic screening, 30 peptides were predicted to have a satisfactory ADMET profile, of which 21 had previously been reported and nine were novel peptides. In order to explore the inhibitory activity of these ligands, authors performed in vitro experiment and peptide Ser-Gly-Ser-Thr had the best IC50 value among the 21 known peptides while Ile-Ile-Tyr and Asn-Pro-Pro-Lys were the best from the derived novel peptides. Furthermore, they carried out molecular docking and dynamic simulation of the two novel compounds to uncover their interaction and binding mechanism. The in silico experiment showed that the two novel peptides were strictly positioned at the active pocket of the S1 and S2 site of ACE and stabilization was facilitated by hydrogen bonds and hydrophobic interactions [61].
In another study, a group of researchers demonstrated the ACE-inhibitory effect of a pentapeptide WPMGF obtained from trypsin hydrolysates of Cyclinasinesis. After their enzymatic kinetic experiment, the Lineweaver Burk plot showed that the peptide competitively inhibits ACE. The authors then performed flexible docking to assess the interaction mechanism of the ligand peptide. Molecular docking simulation revealed that hydrogen bonds, hydrophobic interactions, and coordination bonds were largely responsible for the effective binding of the pentapeptide with ACE [62].
In 2018, Taga et al. [63] in an interesting work assessed the inhibitory potential of X-Hyp-Gly typed peptides on ACE. Their in vitro assay showed that the three peptides Leu-Hyp-Gly, Ile-Hyp-Gly, and Val-Hyp-Gly have strong inhibition against ACE. They also found that Hyp was particularly crucial for the inhibitory activities expressed by the three tripeptides with its replacement with Pro drastically reducing the inhibitory activities of X-Hyp-Gly peptides. This in vitro result was validated with molecular docking with the binding affinity of Leu-Hyp-Gly (-7.7 kcal/mol) higher than the Leu-Pro-Gly mutant derivative (-7.1 kcal/mol).
In another study, Ali et al. [64] examined the binding and inhibitory mechanism of the decapeptide LVV-hemorphin 7 with three different molecular targets, including ACE. They studied mammalian and camel LVV-hemorphin 7 and used in silico methods, such as docking and MM/GBSA to evaluate the hemorphin peptide affinities and MD simulation was used to explore their stabilities with the biological targets. Molecular docking and MMGBSA results showed that camel hemorphin had a stronger binding affinity (-12.31 and -151.57 kcal/mol) with ACE compared to the non-camel type (-10.66 and -119.32 kcal/mol). The RMSD graph also revealed higher stability with ACE for camel (2.5Å) than the mammalian one (3Å) throughout the simulation process. In another research, Lin et al. hydrolyzed Qula casein derived from yak milk casein and predicted high ACE inhibitory activity peptides using a quantitative structure-activity relationship (QSAR) and molecular docking study. They selected 16 peptides for molecular docking on the basis of QSAR modeling. The docking simulation showed that four peptides (KFPQY, MPFPKYP, MFPPQ, and QWQVL) were positioned in the active site of ACE with KFPQY having the highest binding affinity (-10.7 kcal/mol). Furthermore, in order to assess their inhibitory potential in vitro, they synthesized these four novel peptides and the result was found to be in full agreement with the in silico experiment with KFPQY showing the best IC50 value [65].
Finally, Chamata et al. [66] examined the potential interactions and ACE inhibitory activities of peptides synthesized from whey proteins with a molecular docking experiment. The molecular docking result revealed that peptides IIAE, LIVTQ, and LVYPFP formed strong H-bonds with the residues Gln 259, His 331, and Thr 358 in the active pocket of the human ACE (PDB Id: 6F9V). Similarly, the same amino acids were found to interact and form hydrogen bonds with the clinically approved ACE drug (Sampatrilat). Moreover, peptides IIAE and LVYPFP bound with the amino-acid residues Gln259 and His331 of the receptor, respectively, an interaction similar to the binding of other clinically approved drugs (Lisinopril, Captopril, and Elanapril). These findings suggest that these peptides could modulate the activity of ACE, which could further be tested using in vivo studies.
There are also some reports on the discovery of non-peptide ACE inhibitors obtained from plant products. For instance, Muhammad et al. [67] analyzed the inhibitory activity of quercetin glycoside isolated from onions and buckwheat using the in silico docking simulation. The crystal structure of ACE with the PDB ID 1086 was identified as the molecular target for the docking protocol, and Enalapril, a known brand of ACE inhibitor, was used as the standard for comparative study. Molecular docking with PYRX revealed significant binding affinity (-8.5 kcal/mol) of quercetin glycoside compared to the standard (-7.0 kcal/mol). This result showed the potential of the hit compound as a future ACE drug. In another study, Arya et al. studied the molecular mode of interaction of chemical constituents of Clerodendrum colebrookianum against three anti-hypertensive molecular target Rho-associated coiled-coil protein kinase (ROCK), ACE, and phosphodiesterase 5 (PDE5) with molecular docking and MD simulation. They reported that two compounds acteoside and osmanthuside B6 are the most druggable phytocompounds of ACE in the active pocket out of 21 reported compounds from the plant with glide scores of -12.97 and -9.28 kcal/mol. Furthermore, they performed an MD simulation of 20 ns and the result showed that both compounds have good RMSD and RMSF values that accounted for their stability [68].
Meta-analysis and ADMET profiling of hit compounds
In silico screening of drug candidates with ADMET is a vital component in the pharmaceutical R&D industry. The primary reason for preclinical pharmacokinetic and pharmacodynamic consideration is to filter out incompetent and weak drug candidates before advancing to clinical trials. Studies have revealed that undesired ADMET properties of drug candidates are the primary reason for their failure in a clinical trial [69]. In 1993, statistics revealed that 40% of lead compounds failed at the level of clinical trials mainly because of poor pharmacokinetics and bioavailability properties [70]. However, with the advancement in sophisticated technological tools, the use of ADMET for the proper prediction of pharmacokinetic and toxicological parameters has been introduced to screen compounds. This has led to the drastic reduction of drug failure rate at the clinical level from 40% to 11% [71]. Hence, it is imperative that the ADMET parameters of drugs be considered during the preclinical screening of potential lead compounds to avoid failure of drug candidates in clinical trials.
In this review, it is noteworthy that most authors do not put the ADMET profile of drugs into consideration, especially with the reported ACE inhibitors. Only a few following studies where ADMET screening of potential lead compounds of α-glucosidase was considered were found: Mollica et al. in 2017 [48], Meena et al. in 2018 [51], and Laoud et al. in 2017 [47] (Table 1). Only Yu et al. in 2018 [57] reported ACE inhibitors, in which ADMET profiling of hit compounds was considered (Table 1).
.jpg)

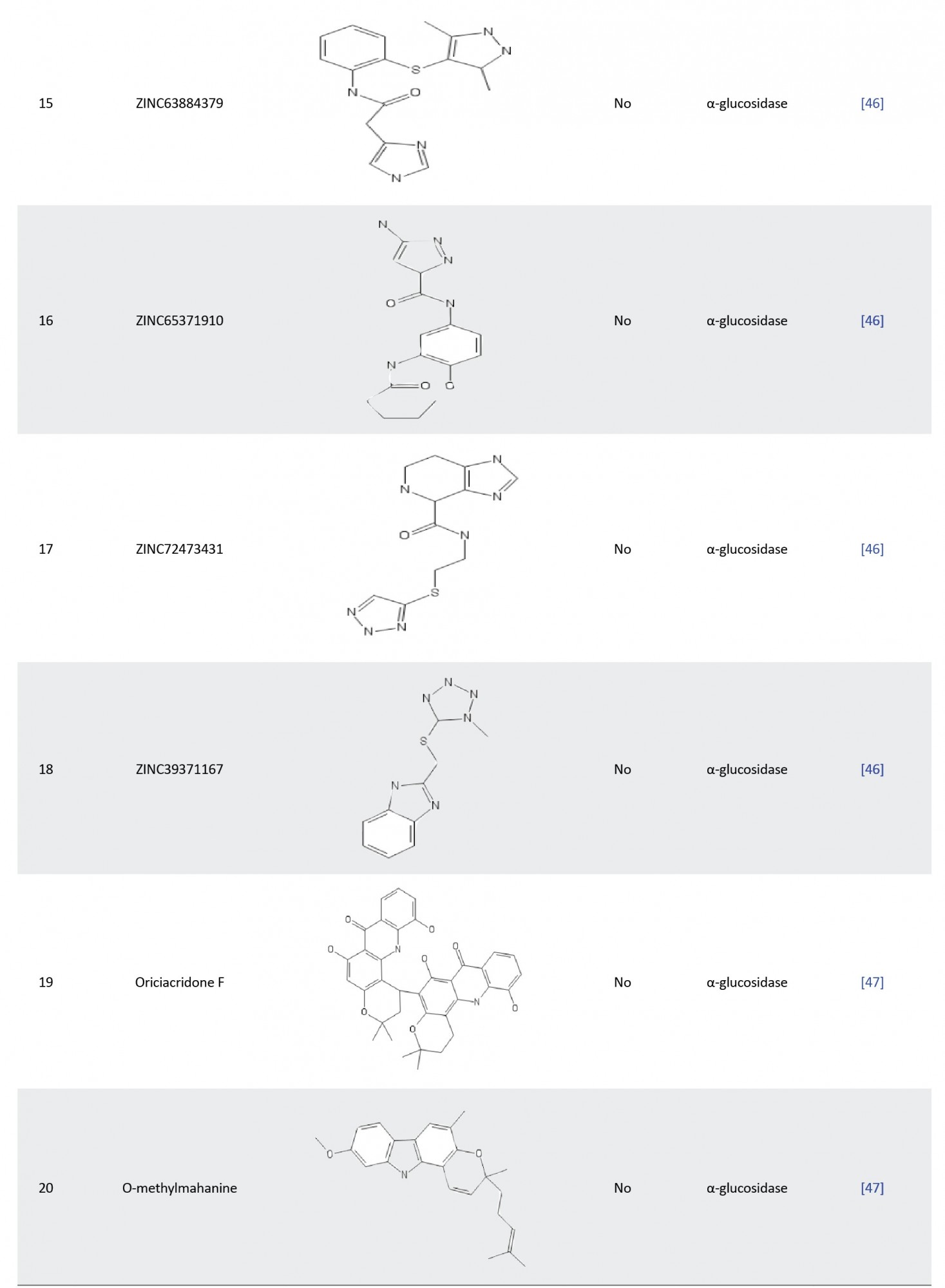


Hence, we performed an in silico meta-analysis of the ADMET profile of the reported drug candidates of α-glucosidase and ACE (Table 2) to weed out the most probable candidates that could fail the clinical trial stage.
Materials and Methods
The SMILE format of 27 reported small molecule inhibitors of α-glucosidase and ACE were retrieved from the PubChem database [72] and deposited to Admetsar 2.0 webserver to evaluate their pharmacokinetic and toxicological properties [73]. The toxicity profiles of peptides were determined using the Toxinpred web server [74].
Results and Discussion
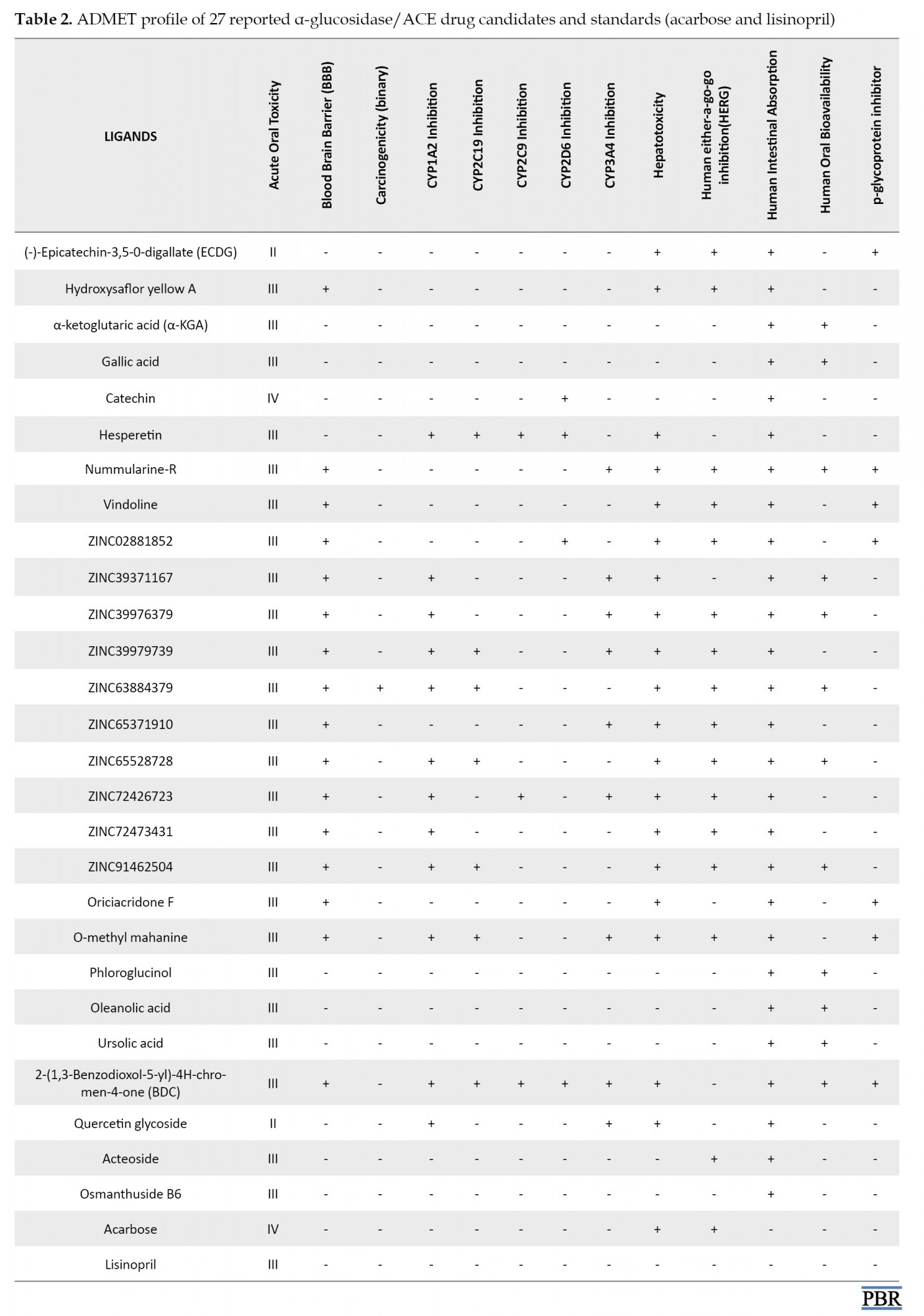
Table 2 depicts the ADMET profile of 27 small molecule inhibitors predicted by the Admetsar2 webserver. In silico ADMET analysis of drugs aids the pharmacokinetics and pharmacodynamics prediction of drug candidates before they can be subjected to further experimental procedures [75]. The underlying reason and ultimate goal for all ADME studies are to properly understand an inhibitor’s metabolite-mediated toxicity and safety properties to make a concrete decision on whether the drug candidates could advance to late-stage preclinical and clinical trials in order to enable filing for an investigational new drug developmental process. In Table 2, the absorption profile shows that all 27 compounds have positive human intestinal absorption (HIA+), meaning that they could be well absorbed in the human intestine. The blood-brain barrier and p-glycoprotein parameters account for the distribution of drug candidates. As depicted in our results (Table 2), most of the compounds show a positive blood-brain barrier value. In contrast, others (ECDG, α-KGA, gallic acid, hesperetin, phloroglucinol, oleanolic acid, ursolic acid, quercetin glycoside, acteoside, and osmanthuside B6) show the negative value, which indicates that they might be unable to permeate the BBB and hence the central nervous system is protected from their action. The inhibition of p-glycoprotein could facilitate the transport of drugs in the cell. It is clear from the table that only ECDG, nummularine-R, Vindoline, ZINC028881852, Oriciacridone-F, O-methyl-mahanine, and BDC are inhibitors of P-glycoprotein.
Metabolism of drugs is carried out by the cytochrome p450 superfamily (CYP), and inhibition of these metabolizing enzymes causes drug-drug interactions and consequently can lead to serious adverse effects due to bio-accumulation of the drug or its metabolite [76]. Only hesperetin inhibited all the CYP450 superfamilies (CYP1A2, CYP2C19, CYP2C9, and CYP2D6) except for CYP3A4. The administration of this drug candidate could affect its metabolism and excretion, leading to accumulation. Conversely, ECDG, hydroxysaflor yellow A, α-KGA, gallic acid, phloroglucinol, and oleanolic acid showed excellent metabolic profiles and will be easily excreted from the body.
The toxicity of compounds is a significant and crucial parameter in the process of drug approval. Disappointingly, all reported compounds with the exception of α-KGA, gallic acid, catechin, phloroglucinol, oleanolic acid, ursolic acid, acteoside, and osmanthuside B6 are hepatotoxic. However, it is worthy of note that only ZINC63884379 is carcinogenic, which potentially could cause cancer. The human ether-a-go-go (HERG) is a K+ channel found in the heart muscle and regulates the rhythm of the heart. If a drug inhibits HERG, it could result in cardiac arrhythmia and eventually death [77, 78]. In the ADMET table, α-KGA, gallic acid, catechin, hesperetin, ZINC39371167, phloroglucinol, oleanolic acid, ursolic acid, BDC, Quercetin glycoside, and osmanthuside B6 are the only drug candidates with no inhibition of the HERG; hence, they are less likely to cause cardiac arrhythmia and death. All compounds with the exception of ECDG and quercetin glycoside (II), have good acute oral toxicity values. Table 3 shows the toxicity profile of peptides. Interestingly, our result revealed that all peptides are safe for administration, as none of them was found to be toxic.

It is crystal clear from Table 2 that α-KGA, gallic acid, catechin, oleanolic acid, phloroglucinol, and ursolic acid are the best drug candidates with excellent pharmacokinetic and pharmacodynamic profiles. We also discovered that these α-glucosidase inhibitors depicted better ADMET properties than the clinically approved type 2 anti-diabetic drug (acarbose). Hence, they could represent a better alternative to type 2 anti-diabetic drugs. Among the reported small molecule inhibitors of ACE (Table 1), Acteoside and osmanthuside B6 demonstrated an excellent ADMET profile with similar parameters compared to the standard clinically approved lisinopril drug.
Conclusion
In this review, we summarized recently in silico drug discovery findings on α-glucosidase and ACE inhibitors, which could be used as pharmacological intervention in the treatment/prevention of T2D and diabetic chronic complications (hypertension and diabetic nephropathy), respectively. A total of 57 promising compounds that could potentially inhibit α-glucosidase or ACE have been identified. Most of the examples of α-glucosidase and ACE inhibitors cited are competitive and peptide inhibitors, respectively. Furthermore, our review highlighted that most authors do not consider the pharmacokinetic and pharmacodynamic screening of potential lead compounds, which could be pivotal in determining their safety profile. It is worthy of note that most lead compounds failed to pass through clinical trials because of poor pharmacokinetic and unacceptable toxicological profiles. Hence, we investigated the ADMET profile of 27 reported small molecule inhibitors using in silico meta-analysis. Our results revealed that among the α-glucosidase inhibitors, α-KGA, gallic acid, catechin, oleanolic acid, phloroglucinol, and ursolic acid demonstrated excellent ADMET properties while acteoside and osmanthuside B-6 showed good pharmacokinetic and pharmacodynamic properties as compounds with ACE inhibitory prowess. In addition, we evaluated the toxicity profile of the reported peptides and interestingly, all proved to be non-toxic. Moreover, after comparing both sets of inhibitors with clinically approved drugs (acarbose and lisinopril), we found them to have better or similar ADMET profiles, suggesting that they could be used as a better alternative in the management of T2D and its complications. Finally, we propose that further studies be conducted on these potential lead compounds to demonstrate their clinical relevance.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Adelusi Temitope Isaac, Oyedele Abdul-Quddus Kehinde; Writing: Oyedele Abdul-Quddus Kehinde, Boyenle Ibrahim Damilare, Adelusi Temitope Isaac, Abdeen Tunde Ogunlana; Review: Boyenle Ibrahim Damilare, Adelusi Temitope Isaac. Editing: Ashiru Mojeed Ayoola, Atanda Opeyemi Emmanuel, Oyedele Abdul-Quddus Kehinde.
Conflict of interest
The authors declared no conflict of interest.
References
The aim of this review was to elucidate the pathophysiological relationship between type 2 DM (T2D) and hypertension and then summarize several studies conducted on in silico screening of α-glucosidase inhibitors in recent years with the objective of recommending them for further preclinical and clinical studies. We further explained the influence of α-glucosidase competitive and allosteric inhibitors on the treatment/management of diabetes and its downstream complications. In addition, since diabetic patients have been found to suffer from chronic complications, such as hypertension and diabetic nephropathy, we also investigated recent computational studies on angiotensin-converting enzyme (ACE) since inhibitors of this enzyme could synergistically prevent hypertension and progression of diabetic nephropathy. Moreover, we noticed that most authors failed to perform in silico ADMET studies of the hit compounds, which is essential in the early phase determination of the safety profile of drug candidateFs and filtering out leads with no favorable pharmacokinetics before they could be subjected to further preclinical ADMET studies. Hence, we carried out in silico meta-analysis on the ADMET profile of the reported compounds and recommend them for further preclinical trials.
The α-glucosidase enzyme has been identified as an efficient molecular target in treating T2D. T2D, also referred to as non-insulin-dependent diabetes, occurs in more than 90% of diabetic patients, and about 80% of them are usually obese [1]. Non-insulin-dependent diabetes mellitus poses a significant threat to the health of humans as a result of its higher rate of occurrence, chronic implication, and complications [2]. Statistical data from the World Health Organization (WHO) highlighted the prevalence of T2DType. In 1980, statistics revealed that over 4.7% of the world population had T2D, and a progressive rise occurred in 2014 after a study showed the percentage had increased to about 8.5% of the world population [3].
Also, α-glucosidase is a key enzyme in carbohydrate digestion responsible for the breakdown of maltose with the concomitant release of glucose [4]. This digestive enzyme acts on the α-1,4 glycosidic bond of disaccharide, a different enzyme from β-glucosidase [5]. Hence, identifying and targeting this digestive enzyme for inhibition to reduce the rate, at which glucose is absorbed in the intestine could be an important therapeutic measure in managing T2D [6].
Several clinically approved drugs against α-glucosidase, such as acarbose, miglitol, and voglibose have been used for the treatment of T2D [4]. These drugs have been designed to act as antidiabetic agents inhibiting the digestion of carbohydrates and slowing down the absorption of glucose. Consequently, this allows the blood glucose level to be maintained constant. Although some clinically approved drugs have been reported against α-glucosidase, these standard drugs are associated with undesirable side effects. Sheliya et al. in 2015 reported that the use of acarbose-a clinically and the widely used antidiabetic drug are associated with several side effects, such as abdominal swelling, pneumatosis cystoides, diarrhea, and flatulence [7]. Many studies have been conducted recently to identify new drug candidates from plant extracts as an alternative to the existing standard drugs with α-glucosidase inhibitory activity [8]. Pharmacological agents derived from plant extracts are increasingly gaining acceptance in the management of diabetes because they are easily accessible and do not require laborious pharmaceutical synthesis compared to their synthetic counterparts [9].
Also, it has been reported that more than 75% of patients with diabetes suffer from microvascular complications, such as hypertension [10], and need antihypertensive drugs, such as ACE inhibitors (ACEI) to treat their condition. Moreover, aside from the antihypertensive effect of this drug, they also help delay the progression of nephropathy in diabetic patients who are normotensive and hypertensive. In addition, 25-40% with type 1 or 2 diabetes have diabetic nephropathy after 20-25 years of the onset of the disease [11]. The distinguishing clinical characteristics of diabetic nephropathy involve a gradual increase in the level of albuminuria and a declined glomerular filtration rate (GFR), which occur with an accompanying increase in blood pressure and eventually cause end-stage renal failure [12]. Angiotensin-2, a potent vasoconstrictor produced as a result of ACE activity, causes diabetic nephropathy by inducing hypertrophy in mesangial and epithelial cells [13] and also promoting the production of transforming growth factor (TGF-β), which has been identified as a causative agent for glomerulosclerosis [14]. ACEIs are the most widely used agents in diabetic patients due to their capacity to slow the deterioration of renal function and decrease proteinuria [15]. In a comprehensive review by some group of scientists, findings support the use of ACE inhibitors in patients with diabetes mellitus and hypertension because aside from its antihypertensive property, it also prevents chronic complications, such as diabetic nephropathy as a result of its renoprotective effect [16].
Pathophysiological interplay between T2D, hypertension, and diabetic nephropathy
Diabetes, most especially T2D represents a global health problem of epidemic proportions and arises as a result of insulin resistance [17]. Enzymes responsible for the digestion of carbohydrates have been recognized as an important target in the management of T2D. After the ingestion of carbohydrates, the digestive process of polysaccharides begins with a breakdown into oligosaccharides by salivary α-amylase and subsequently degraded into disaccharides with pancreatic α-amylase. The α-amylase enzyme hydrolyzes the α-bonds of large α-linked carbohydrates and is found mostly in saliva and pancreatic juice [18]. Disaccharides in the small intestine are catabolized to monosaccharides, such as glucose, fructose, etc. by α-glucosidase produced in the intestinal epithelial cells. Hence, the key enzyme responsible for the final breakdown of polysaccharides to glucose is α-glucosidase. Studies have shown that inhibitors of α-glucosidase could retard the uptake of glucose, decrease postprandial hyperglycemia, and consequently be useful in the management of T2D [19, 20].
One of the major vascular complications of T2D is hypertension caused by high blood pressure. Also, 70% of diabetic patients also develop high blood pressure [10]. It has been suggested that high blood glucose levels in the bloodstream could enhance the nonenzymatic glycosylation of several molecules and stimulate the production of reactive oxygen species (ROS), which may overwhelm the body’s antioxidant system and subsequent manifestation of diabetic chronic complications [21]. Recently, Marc et al. reported that blindness, renal disease, and cardiovascular disease are the chronic complication of diabetes [22]. Among the risk factors of cardiovascular disease is hypertension, which is approximately twice as frequent in diabetic patients than in those without the disease [23]. Hence, regulating postprandial hyperglycemia is an important therapeutic strategy in the management or treatment of diabetes and synergistically could enhance a holistic decrease in high blood pressure related to diabetes as shown in Figure 1 [24].
Also, the administrations of pharmacological agents inhibiting the renin-angiotensin-aldosterone system (RAAS) have been reported to hinder the onset of T2D in high-risk populations [25, 26]. In the renin angiotensin aldosterone system, the hepatic angiotensinogen is cleaved by renal synthesized renin to produce Ang I, which is then converted to a strong vasoconstrictor-Ang II in the lung by the action of ACE and thereby causing high blood pressure. Although the use of a pharmacological intervention that causes blockage of the RAAS pathway to reduce high blood pressure in subjects with hypertension, some reports obtained from rodents and cell experiments suggest that ameliorating insulin sensitivity by improving the function of adipose tissue and enhancing β-cell function through improved islet perfusion and anti-fibrotic effect are the proposed mechanism of action, by which therapeutic agents blocking RAAS pathway could reduce the risk of T2D [27-30]. The administration of angiotensin-converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) blocking the RAAS system reduced the risk of T2D in hypertensive patients [31-33].
T2D and hypertension caused by hyperglycemia and high blood pressure respectively have also been linked to diabetic nephropathy (DN). High blood glucose could lead to the formation of Amadori products (altered proteins) and advanced glycation end products, which are part of the component of DN. Furthermore, activation of the electron transport chain caused by hyperglycemia can lead to increased generation of ROS, which may be the initiating agent in the development of diabetic complications. The hemodynamic pathway involving the formation of growth factors, hypertrophy, extracellular matrix accumulation, cytokines, proteinuria, and ROS has been implicated in the progression of DN [34]. Hyperglycemia, advanced glycation end products (AGE), and ROS could work in harmony to cause the release of cytokines and growth factors through a signal transduction pathway that involves protein kinase C and nuclear transcription factor (NF-KB). Transforming growth factor (TGF-β) could induce hypertrophy of the kidney cells and accumulation of extracellular matrix proteins [35]. Similarly, angiotensin II could induce hypertrophy in mesangial and epithelial cells [13], and enhance the formation of TGF-β, which are part of the key players in DN [14]. In addition, studies have shown that the strong vasoconstrictor Ang II produced as a result of the activation of the renin-angiotensin aldosterone system is involved in almost all stages of the development of DN [36]. The emergence of hypertension in a diabetic patient could increase the risk of developing end-stage renal disease five to six folds compared to patients who are hypertensive alone [37]. Inhibitors of the renin-angiotensin system, such as ACEI and angiotensin receptor blockers (ARB) have been shown to impede progressive kidney impairment and prevent the onset of end-stage kidney disease in patients with DN [38].
Competitive inhibitors of α-glucosidase
The experimental x-ray crystallography technique has revealed the architectural structure of α-glucosidase, which consists of N- and C-terminal catalytic domains [39, 40]. Researchers have thus identified the active residues in the catalytic domain of this enzyme and labeled them as a therapeutic target for the design of antidiabetic agents in the drug discovery process. The vast majority of reports on the screening of α-glucosidase inhibitors that use the molecular modeling approach are focused on the recognition of small molecules derived from natural plant extracts acting as competitive inhibitors or blockers of the enzyme’s active site.
A good example of a competitive inhibitor of α-glucosidase was reflected in the work where Epicatechin-3,5-O-gallate (ECDG) derived from Orostachys japonicus binds to the receptor active site [8]. A number of compounds derived from Orostachys japonicus were docked against the α-glucosidase binding site; however, ECDG proved to be the best inhibitor of the enzyme with autodock score of -10.17 kcal/mol. The major interaction contributing to this high binding affinity was nine hydrogen bonds with six amino acids, including Pro224, Gln233, Asn236, His240, Asn241, and Pro304. The molecular docking result suggests that ECDG may inhibit the catalytic reaction of α-glucosidase by covering the active site pocket. Molecular dynamics of 10 ns were also executed to uncover the behavior and dynamics of this receptor-ligand binding process. The molecular dynamic result showed that ECDG mainly had 1-3 hydrogen bonds, with receptor residues in the stable complex. Asn236 was found to be a critical residue as it participated in the formation of hydrogen bonds throughout the simulation. The root means square deviation (RMSD) was evaluated as 0.30 nm after 10 ns, which represents the stability of the complex. This result revealed that ECDG may be a potential drug candidate for α-glucosidase, which could be used in the treatment of T2D.
Xu et al. [41] demonstrated the inactivation of α-glucosidase with Hydroxysaflor yellow A (HSYA) using a molecular dynamics study. The targeted receptor and HSYA extracted from Carthamus tintorius were used in the docking experiment to obtain the best ligand pose. The result was then used to conduct a molecular dynamic simulation of 10 ns, which indicated the active site interaction of HSYA with G217, A278, H279, and G280. The molecular modeling result was shown to be consistent with their enzyme kinetics study, which proves that HSYA might interrupt the substrate binding activity of α-glucosidase in a competitive manner and hence acts as a potential anti-diabetic agent in the treatment of T2D.
Xiong et al. [42] assessed the inhibitory effect of α-ketoglutaric acid (AKG) on α-glucosidase. The binding pocket of α-glucosidase was assessed for AKG docking with α-glucosidase-like-proteins, using the sequence alignment program in the HHblits server. VMD was used to determine the position of the bound ligands in the enzyme pocket. The overlapping regions around the ligand-binding points and the α-glucosidase pockets were identified to define the AKG binding site. The molecular docking process was carried out using AutoDock Vina version 1.1.2 and the result showed that AKG was stably positioned at the existing pocket of the enzyme. They also performed a molecular dynamic simulation of 10 ns, which reveals that TYR313, ASN412, and ARG439 formed hydrogen bonds with AKG and thereby facilitating its binding and stability. They validated this molecular modeling result with an experimental enzyme kinetic study, which shows that AKG competitively inhibits α-glucosidase.
In 2019, Choudhary DK, et al. [43] examined the inhibitory activity and mechanism of polyphenols extracted from fava bean on α-glucosidase. They performed molecular docking on all the extracted polyphenols and discovered that only two compounds (gallic acid and catechin) had the highest binding affinity (-6.58 and -7.25 kcal/mol, respectively) in comparison with the standard acarbose drug (-6.35 kcal/mol). Furthermore, both compounds were found to have four hydrogen bonds and the critical amino acid interaction responsible for their binding affinity was located at the active site of the enzyme with Asn324, Asn326, and Arg197 for gallic acid and Asp198, Asn324, Asp326, and Tyr63 for catechin. Molecular dynamics with GROMACS was used to run a simulation that spanned 50 ns and the result substantiated the interaction between gal:α-glucosidase and cat:α-glucosidase as stable complexes with the RMSD varying between 0.15 and 0.35 nm throughout the simulation period. These findings revealed that ethanolic seed extract from fava beans could be an efficient α-glucosidase inhibitor.
Gong et al. [4] evaluated the inhibitory effect of hesperetin (a flavonoid derivative) on α-glucosidase based on computational molecular docking and dynamic simulation. The docking experiment showed that hesperetin was tightly bounded and buried into the active pocket of the enzyme with a binding affinity of -7.1 kcal/mol. The docked structure was used to run 10 ns MD simulations to estimate how tightly the ligand binds to α-glucosidase. The MD simulation revealed the critical amino acids responsible for hesperetin: α-glucosidase binding, which were Lys155, Ser156, Phe157, Gly159, Phe231, Gly159, Phe231, Asn241, Ala 278, His279, Glu304, Thr307, Ser308, Pro309, Phe311, and Arg312. After the MD simulation, the distance profile analysis was carried out to determine whether the α-glucosidase pocket was blocked by hesperetin. Interestingly, the distance profile revealed that five groups of hesperetin were found in close contact (short distance below 6Å) with six crucial residues (Lys155, Asn241, Glu304, Pro309, Phe311, and Arg312) in the active site ofα-glucosidase. This analysis showed that hesperetin blocked the binding pocket of the receptor, thereby inhibiting its catalytic activity. The in silico result was also found to be in agreement with their experimental enzyme kinetic studies, which uncovered the binding and inhibitory mechanism of hesperetin as an α-glucosidase competitive inhibitor.
In another study, Rahman et al. [5] investigated the inhibitory potential of 32 isolated alkaloids from plants against α-glucosidase using a molecular docking screening process. The criteria for their selection of these compounds were based on already reported in vitro and in vivo activities of these alkaloids on α-glucosidase inhibition. The docking process was done by the molecular operating environment (MOE) software for the prediction of the binding affinities of the selected 32 alkaloids against α-glucosidase. The results revealed nummularine-R and vindoline as the most potent compounds with docking scores of -14.5691 and -13.2250, respectively. These docking scores were also found to be very close to standard α-glucosidase inhibitors-acarbose and miglitol (-14.7983 and -15.4423, respectively). Furthermore, both ligands recorded good interactions with the active site of the target enzyme as the most potent compound (nummularine-R) formed four hydrogen interactions with Gln121, Met122, Arg331, and Gly546 while the second most potent compound (vindoline) had three hydrogen bonds and one arene-arene interaction with amino acids Ala93, Ala97, Gln121, and Trp126. The results of these docking screenings were consistent with previously reported in vitro antidiabetic experiments [44, 45], which revealed the promising potential of nummularine-R and vindoline as α-glucosidase inhibitors.
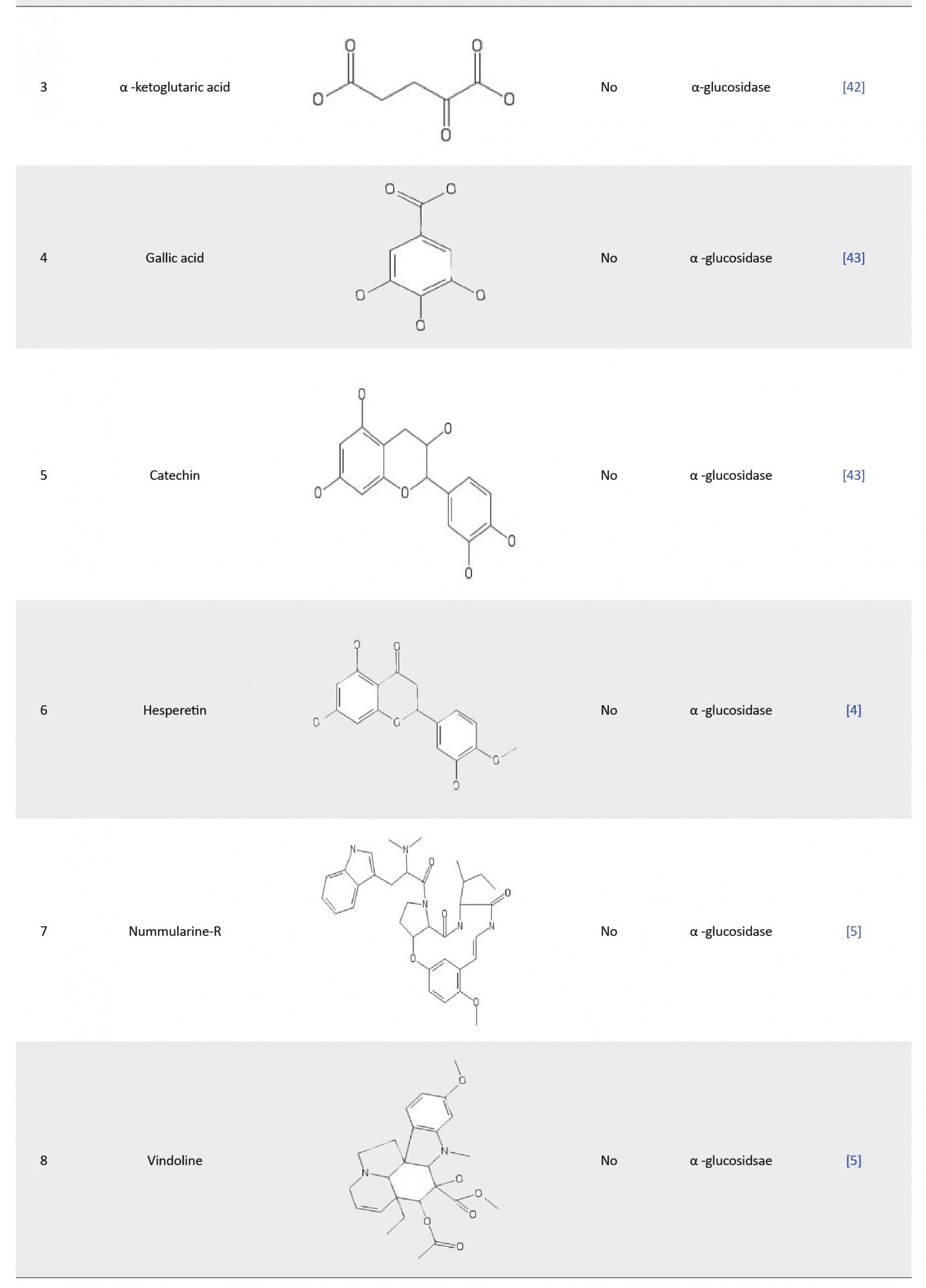
In another study, Laoud et al. identified some novel inhibitors of nt-MGAM using QSAR, docking, and ADME for their virtual screening process. Firstly, they performed molecular docking of a set of salacinol derivatives into the active pockets of N-terminal α-glucosidase, and then the authors used an atom-based QSAR model to identify pharmacophoric features of salacinol derivatives responsible for the enzyme inhibition. Virtual screening was subsequently carried out to screen potential compounds with similar chemical scaffolds in the ZINC database. Eventually, ten hit compounds (ZINC 39979739, ZINC 72426723, ZINC 39976379, ZINC 02881852, ZINC 91462504, ZINC 65528728, ZINC 63884379, ZINC 65371910, ZINC 72473431, and ZINC 39371167) were identified and satisfactory ADME profiles were then obtained for this drug-like compounds, which indicated that the salacinol analogues may be a promising drug in the treatment of T2D [46].
In other research, Zafar et al. examined the α-glucosidase inhibitory potential of some isolated alkaloids. MOE software was used to obtain the tertiary structure of the biological target and molecular docking was used to dock the 37 isolated alkaloids to the modeled protein. The result revealed that compounds 17 (oriciacridone F) and 24 (o-methyl mahanine) possess striking interactions with the active amino acid of the enzyme and the interaction demonstrated were similar to those of the standard drugs (acarbose and miglitol), making them potential lead compound against α-glucosidase [47].
There are also theoretical reports on the use of molecular modeling experiments for the assessment of the inhibitory potential of peptides against α-glucosidase. An example of this could be found in the study by Mollica et al. [48], where they assessed the screening of a combinatorial peptide library that could act as a promising inhibitor of α-glucosidase through binding to the enzyme’s active pocket. Hence, in order to identify novel peptide candidates that could act as potent inhibitors of the molecular target, the top 12 hit compounds were selected for MD simulations from the docking result based on their higher binding affinity. They performed an MD simulation of 20 ns to analyze the stability of the peptides with α-glucosidase. After the simulation, four peptides (H-Phe-Arg-Asn-NH2, H-Trp-Arg-Ala-NH2, H-Lys-Arg-Met-NH2, and H-Asn-Arg-Asn-NH2) were chosen as the final hit compounds on the basis of their lower RMSD value, which signals higher stability compared to other peptides that passed through the MD simulation. Interestingly, the in silico result was also found to be consistent with their in vitro experiment with the four peptides having inhibitory potency that is very similar in relation to standard acarbose. This study showed that peptides could act as potential agents in the treatment of diabetes.
Allosteric inhibitors of α-glucosidase
Targeting the allosteric site of enzymes or proteins may emerge as a compelling alternative inhibition mechanism of some ligands other than binding to the receptor active site and some examples of these inhibitors have been reported in the modulation of α-glucosidase.
Recently, in 2019, Wan et al. [49] investigated the inhibitory potency of phloroglucinol against α-glucosidase using enzyme kinetics and molecular dynamics study. Earlier in their experiment, enzyme kinetic results had shown that phloroglucinol is a reversible inhibitor of α-glucosidase in a dose-dependent but non-competitive mode. Thus, in order to assess this mode of inhibition in silico, they performed molecular docking and MD simulation study on α-glucosidase. The tertiary structure of the enzyme (Saccharomyces cerevisae) was obtained using homology modeling as the x-ray crystallographic structure was unavailable at the time. Based on the predicted phloroglucinol-binding residues (Tyr71, His111, Phe177, Arg212, Asp214, Thr215, Gly217, Leu218, Glu276, His348, and Asp349), AutoDock Vina v.1.1.2 was used for the molecular docking of phloroglucinol to α-glucosidase to obtain the best pose of the protein-ligand complex. The binding affinity of the complex was calculated as -7.48 kcal/mol and the best pose was subjected to MD simulation of 10 ns. The docking and MD simulations revealed that phloroglucinol was almost completely localized in the α-glucosidase binding site while interacting with a single binding site near the active pocket. Furthermore, the phloroglucinol binding was found to be more stable at the entry point of the active site (rather than inside the enzyme’s active pocket) with the distant profile analysis revealing the distance between the interaction of residues and relative bound inhibitor to be less than 6Å. They concluded that phloroglucinol is a promising allosteric inhibitor of α-glucosidase, which could interfere in the normal catalysis of the enzyme and therefore, could be used in the treatment of T2D.
In another study, Ding et al. [50] examined the synergistic inhibition mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Prior to their in silico assessment of the two compounds on α-glucosidase, they carried out an enzyme kinetic inhibition study, which revealed that oleanolic acid and ursolic acid exhibited effective inhibitory activities with IC50 estimated as (6.35±0.02)×10-6 and (1.69±0.03)×10-5 molL-1, respectively in a reversible and non-competitive manner. This result was further validated with their docking experiment, which showed that the two compounds bind to different allosteric sites on α-glucosidase in cavity 2 and cavity 4, respectively. Oleanolic acid interacted with allosteric residues, such as Trp14, Lys12, Ser295, Ala289, His258, Tyr292, Lys262, Val265, Ile271, and Glu270 with -4.63kcal/mol while ursolic acid bound with amino acids Trp465, Glu405, Val407, Lys410, Asn411, Ser179, Arg180, Gln67, Gln66, and Met69 with -4.37 kcal/mol. This protein-ligand interaction does not correspond to the amino acid residues on the active pocket of the enzyme, which were positioned in cavity 1 and thereby allowing allosteric binding to perturb the conformational dynamics of the enzyme leading to a reduction in the catalytic activity of α-glucosidase. The in silico result coupled with in vitro study revealed the inhibitory mechanism of oleanolic acid and ursolic acid on α-glucosidase and their management in the treatment of T2D.
Meena et al. [51] assessed the α-glucosidase inhibitory potential of a synthetic flavone derivative 2-(benzo [d] [1, 3] dioxol-5-yl)-4H-chromen-4-one (BDC) with their in vitro enzyme kinetic and in silico studies. The mode of the mechanism of BDC had been earlier revealed by authors in their in vitro experiment as non-competitive with the maximum α-glucosidase inhibitory potential of the ligand 22.4 folds over the maximum competitive inhibition depicted by standard acarbose drug (27.6 to 669.57 μM). In other to substantiate and show the allosteric interaction of BDC with α-glucosidase, they performed in silico molecular docking experiment. The active sites of the enzyme were determined using CASTp 3.0 web server.
The docking experiment of acarbose with the receptor revealed that acarbose competes for the active pocket of the α-glucosidase enzyme with a minimum binding affinity of -9.23 kcal/mol. Two hydrogen bonds (LYS156:H23 (2.03Ao) and SER241:HN (2.02Ao)) were recognized as the interaction between the receptor and the ligand while interactions are electrostatic, Van der Waals, and hydrophobic interactions. Contrary to this mode of interaction in acarbose, BDC depicted allosteric binding with its interaction on α-glucosidase found to be different from the active pocket residues. Two hydrogen bonds were also observed between LYS373:H22 (2.077Ao) and LYS568:H23 (2.09Ao) of the protein-ligand complex while other interactions are electrostatic/hydrophobic bonds. The binding of BDC [-8.64kcal/mol) showed that the potential drug candidate inhibitory effect was significant compared to the standard clinically approved acarbose (-9.23 kcal/mol). Furthermore, Lipinski’s rule of five was used to evaluate the drug-likeness profile of the ligand and by obeying all rules coupled with its high binding affinity with α-glucosidase, the authors concluded that BDC could be a promising lead compound in the management of diabetes.
Angiotensin-converting enzyme (ACE) structural information, inhibitors, and molecular modeling
Investigation of the molecular structure of ACE has revealed that the human angiotensin-converting enzyme comprises two isoforms: somatic ACE, which consists of two catalytic homologous domains (N- and C-terminal with 60% similarities) [52], and testis ACE, which has only one domain that is structurally similar to the C-terminal catalytic domain of somatic ACE [53]. The two homologous domains of ACE could hydrolyze angiotensin-1; however, both have different pharmacological potentials as they have different substrate specificities. Fuchs et al. revealed that C-domain is enough to regulate blood pressure in vivo [54] and is thus a major site for angiotensin-2 production. Conversely, the N-domain is more specific for the control of haemopoietic stem cell proliferation and differentiation due to its breakdown of anti-fibrotic haemoregulatory peptide AcSDKP [55, 56]. Hence, it is important that the C-domain of ACE be given more preference in the selection of a target for ACE inhibitors during the rational drug discovery process.
Computational molecular modeling technique has been used in the screening of ACE inhibitors. Most of the investigations on the computational screening of ACE inhibitors have focused on the recognition of peptides gotten from hydrolysates of protein or natural sources. For example, Yu et al. [57] explored the ACE-inhibitory potential of some selected tripeptides obtained from in silico gastrointestinal proteolytic cleavage of Larimichthys crocea nebulin protein. The 168 unreported tripeptides derived from the proteolytic process were subjected to drug-likeness and ADME screening. Eventually, four tripeptides (EGF, MDY, GDL, and HGR) were chosen as they have reasonable molecular weight and better pharmacokinetic profiles, such as human intestinal absorption, water solubility, and CYP450 inhibition compared to others. Hence, these four peptides were selected for molecular docking experiments. The crystal structure of ACE in complex with lisinopril (1o86) was retrieved from the protein data bank and the authors performed the docking protocols with CDOCKER PROGRAM. The binding affinity of -92.4782, -90.8416, -80.8434, and -83.4035 kcal/mol was obtained for HGR, EGF, GDL, and MDY tripeptides, respectively. On the basis of binding affinity (-92.4782 kcal/mol) and the number of intermolecular hydrogen bonds (14 H-bond), Yu et al. selected HGR as the best ACE inhibitor and subjected it to further in vitro experiments.
In another study, De Oliveira et al. demonstrated the ACE-inhibitory mechanism of six peptides (ALNEINQFYQK, NAVPITPTLNR, FALPQYLK, FFVAPFPEVFGK, YLGYEQLLR, and HQGLPQEVLNENLLR) derived from hydrolysis of milk caseins. The crystal structure of human ACE (pdb id:1o86) was obtained from the RCSB protein data bank and used as a molecular target for the docking process. De Oliveira et al. performed a docking experiment of the peptide ligands to the active site of the N- and C-catalytic domains of ACE using AutodockVina. They reported that out of the entire 12 docked complexes, only peptide FALPQYLK N-oriented pose failed to bind with ACE while others recorded good interaction with the enzyme active site. In order to examine the dynamics of these complexes, they performed a molecular dynamic simulation of 50 ns using GROMACS version 5.1.4. According to RMSD analysis (less than 3Å), no noticeable conformational change of all the complexes was observed throughout the simulation with respect to their initial docked pose. The RMSF analysis of their in silico study showed that some amino acids located around the ACE extremities have high fluctuation regardless of the ligand geometry or conformation and do not inhibit ACE. They then proposed that this finding could give insight into a computational prediction of promising and non-promising ACE inhibitory peptides obtained from other sources [58].
In another research, Tahir et al. [59] assessed the ACE inhibitory potential of some peptides derived from a blood pressure-regulating honey protein (MRJPI). I-TASSER homology modeling server was used to determine the tertiary structure of MRJPI. However, in order to improve the quality of the modeled structure, optimization of the receptor was carried out with molecular dynamics of 30ns. The RMSD, RMSF, and B-factor analysis revealed the structural stability of MRJPI and was considered to be suitable for further in silico assessment. To identify potential inhibitory peptides, an AHTpin server based on a scoring vector machine of regression models was used for the proteolysis of the optimized protein. They then performed a docking study of all derived peptides to evaluate their binding affinity with ACE. Tahir et al. then identified the hit ligand “EALPHVPIFDR” that had the highest ACE-inhibitory potential from its binding affinity value (-58.29 kcal/mol). Interestingly, from the knowledge of the peptides-ACE interaction, the researchers discovered that Tyr residue was particularly a crucial residue, which could be a therapeutic target during ACE-inhibition rational drug design process.
In another study, Auwal et al. [60] examined the ACE inhibitory potential of five peptides (Ala-Leu-Gly-Pro-Gln-Phe-Tyr, Lys-Val-Pro-Pro-Lys-Ala, Leu-Ala-Pro-Pro-Thr-Met, and Asp-Val-Leu-Ile-Gln) derived from stone fish protein hydrolysate. Their in vitro studies had shown that the five peptides are promising ACE inhibitors with satisfactory IC50 values. Hence, in order to substantiate this finding, the authors performed a molecular docking experiment of the potential drug candidates with Glide XP software. Interestingly, Auwal et al. docking result was found to be consistent with their in vitro experiment with peptide sequence Ala-Leu-Gly-Pro-Gln-Phe-Tyr showing the highest binding affinity (-10.374 kcal/mol) and lowest IC50 value. Likewise, the Gly-His-Pro-Val-Leu sequence had the lowest inhibitory effect both in silico (-8.635 kcal/mol) and in vitro levels. In this study, the scientists demonstrated the effectiveness and promising inhibitory activities of a peptide derived from stone fish. In another research, a group of researchers derived 126 potential ACE-inhibitory peptides from Tobarodespacifus using in silico and bioinformatic tools. After their pharmacokinetic and pharmacodynamic screening, 30 peptides were predicted to have a satisfactory ADMET profile, of which 21 had previously been reported and nine were novel peptides. In order to explore the inhibitory activity of these ligands, authors performed in vitro experiment and peptide Ser-Gly-Ser-Thr had the best IC50 value among the 21 known peptides while Ile-Ile-Tyr and Asn-Pro-Pro-Lys were the best from the derived novel peptides. Furthermore, they carried out molecular docking and dynamic simulation of the two novel compounds to uncover their interaction and binding mechanism. The in silico experiment showed that the two novel peptides were strictly positioned at the active pocket of the S1 and S2 site of ACE and stabilization was facilitated by hydrogen bonds and hydrophobic interactions [61].
In another study, a group of researchers demonstrated the ACE-inhibitory effect of a pentapeptide WPMGF obtained from trypsin hydrolysates of Cyclinasinesis. After their enzymatic kinetic experiment, the Lineweaver Burk plot showed that the peptide competitively inhibits ACE. The authors then performed flexible docking to assess the interaction mechanism of the ligand peptide. Molecular docking simulation revealed that hydrogen bonds, hydrophobic interactions, and coordination bonds were largely responsible for the effective binding of the pentapeptide with ACE [62].
In 2018, Taga et al. [63] in an interesting work assessed the inhibitory potential of X-Hyp-Gly typed peptides on ACE. Their in vitro assay showed that the three peptides Leu-Hyp-Gly, Ile-Hyp-Gly, and Val-Hyp-Gly have strong inhibition against ACE. They also found that Hyp was particularly crucial for the inhibitory activities expressed by the three tripeptides with its replacement with Pro drastically reducing the inhibitory activities of X-Hyp-Gly peptides. This in vitro result was validated with molecular docking with the binding affinity of Leu-Hyp-Gly (-7.7 kcal/mol) higher than the Leu-Pro-Gly mutant derivative (-7.1 kcal/mol).
In another study, Ali et al. [64] examined the binding and inhibitory mechanism of the decapeptide LVV-hemorphin 7 with three different molecular targets, including ACE. They studied mammalian and camel LVV-hemorphin 7 and used in silico methods, such as docking and MM/GBSA to evaluate the hemorphin peptide affinities and MD simulation was used to explore their stabilities with the biological targets. Molecular docking and MMGBSA results showed that camel hemorphin had a stronger binding affinity (-12.31 and -151.57 kcal/mol) with ACE compared to the non-camel type (-10.66 and -119.32 kcal/mol). The RMSD graph also revealed higher stability with ACE for camel (2.5Å) than the mammalian one (3Å) throughout the simulation process. In another research, Lin et al. hydrolyzed Qula casein derived from yak milk casein and predicted high ACE inhibitory activity peptides using a quantitative structure-activity relationship (QSAR) and molecular docking study. They selected 16 peptides for molecular docking on the basis of QSAR modeling. The docking simulation showed that four peptides (KFPQY, MPFPKYP, MFPPQ, and QWQVL) were positioned in the active site of ACE with KFPQY having the highest binding affinity (-10.7 kcal/mol). Furthermore, in order to assess their inhibitory potential in vitro, they synthesized these four novel peptides and the result was found to be in full agreement with the in silico experiment with KFPQY showing the best IC50 value [65].
Finally, Chamata et al. [66] examined the potential interactions and ACE inhibitory activities of peptides synthesized from whey proteins with a molecular docking experiment. The molecular docking result revealed that peptides IIAE, LIVTQ, and LVYPFP formed strong H-bonds with the residues Gln 259, His 331, and Thr 358 in the active pocket of the human ACE (PDB Id: 6F9V). Similarly, the same amino acids were found to interact and form hydrogen bonds with the clinically approved ACE drug (Sampatrilat). Moreover, peptides IIAE and LVYPFP bound with the amino-acid residues Gln259 and His331 of the receptor, respectively, an interaction similar to the binding of other clinically approved drugs (Lisinopril, Captopril, and Elanapril). These findings suggest that these peptides could modulate the activity of ACE, which could further be tested using in vivo studies.
There are also some reports on the discovery of non-peptide ACE inhibitors obtained from plant products. For instance, Muhammad et al. [67] analyzed the inhibitory activity of quercetin glycoside isolated from onions and buckwheat using the in silico docking simulation. The crystal structure of ACE with the PDB ID 1086 was identified as the molecular target for the docking protocol, and Enalapril, a known brand of ACE inhibitor, was used as the standard for comparative study. Molecular docking with PYRX revealed significant binding affinity (-8.5 kcal/mol) of quercetin glycoside compared to the standard (-7.0 kcal/mol). This result showed the potential of the hit compound as a future ACE drug. In another study, Arya et al. studied the molecular mode of interaction of chemical constituents of Clerodendrum colebrookianum against three anti-hypertensive molecular target Rho-associated coiled-coil protein kinase (ROCK), ACE, and phosphodiesterase 5 (PDE5) with molecular docking and MD simulation. They reported that two compounds acteoside and osmanthuside B6 are the most druggable phytocompounds of ACE in the active pocket out of 21 reported compounds from the plant with glide scores of -12.97 and -9.28 kcal/mol. Furthermore, they performed an MD simulation of 20 ns and the result showed that both compounds have good RMSD and RMSF values that accounted for their stability [68].
Meta-analysis and ADMET profiling of hit compounds
In silico screening of drug candidates with ADMET is a vital component in the pharmaceutical R&D industry. The primary reason for preclinical pharmacokinetic and pharmacodynamic consideration is to filter out incompetent and weak drug candidates before advancing to clinical trials. Studies have revealed that undesired ADMET properties of drug candidates are the primary reason for their failure in a clinical trial [69]. In 1993, statistics revealed that 40% of lead compounds failed at the level of clinical trials mainly because of poor pharmacokinetics and bioavailability properties [70]. However, with the advancement in sophisticated technological tools, the use of ADMET for the proper prediction of pharmacokinetic and toxicological parameters has been introduced to screen compounds. This has led to the drastic reduction of drug failure rate at the clinical level from 40% to 11% [71]. Hence, it is imperative that the ADMET parameters of drugs be considered during the preclinical screening of potential lead compounds to avoid failure of drug candidates in clinical trials.
In this review, it is noteworthy that most authors do not put the ADMET profile of drugs into consideration, especially with the reported ACE inhibitors. Only a few following studies where ADMET screening of potential lead compounds of α-glucosidase was considered were found: Mollica et al. in 2017 [48], Meena et al. in 2018 [51], and Laoud et al. in 2017 [47] (Table 1). Only Yu et al. in 2018 [57] reported ACE inhibitors, in which ADMET profiling of hit compounds was considered (Table 1).
Hence, we performed an in silico meta-analysis of the ADMET profile of the reported drug candidates of α-glucosidase and ACE (Table 2) to weed out the most probable candidates that could fail the clinical trial stage.
Materials and Methods
The SMILE format of 27 reported small molecule inhibitors of α-glucosidase and ACE were retrieved from the PubChem database [72] and deposited to Admetsar 2.0 webserver to evaluate their pharmacokinetic and toxicological properties [73]. The toxicity profiles of peptides were determined using the Toxinpred web server [74].
Results and Discussion
Table 2 depicts the ADMET profile of 27 small molecule inhibitors predicted by the Admetsar2 webserver. In silico ADMET analysis of drugs aids the pharmacokinetics and pharmacodynamics prediction of drug candidates before they can be subjected to further experimental procedures [75]. The underlying reason and ultimate goal for all ADME studies are to properly understand an inhibitor’s metabolite-mediated toxicity and safety properties to make a concrete decision on whether the drug candidates could advance to late-stage preclinical and clinical trials in order to enable filing for an investigational new drug developmental process. In Table 2, the absorption profile shows that all 27 compounds have positive human intestinal absorption (HIA+), meaning that they could be well absorbed in the human intestine. The blood-brain barrier and p-glycoprotein parameters account for the distribution of drug candidates. As depicted in our results (Table 2), most of the compounds show a positive blood-brain barrier value. In contrast, others (ECDG, α-KGA, gallic acid, hesperetin, phloroglucinol, oleanolic acid, ursolic acid, quercetin glycoside, acteoside, and osmanthuside B6) show the negative value, which indicates that they might be unable to permeate the BBB and hence the central nervous system is protected from their action. The inhibition of p-glycoprotein could facilitate the transport of drugs in the cell. It is clear from the table that only ECDG, nummularine-R, Vindoline, ZINC028881852, Oriciacridone-F, O-methyl-mahanine, and BDC are inhibitors of P-glycoprotein.
Metabolism of drugs is carried out by the cytochrome p450 superfamily (CYP), and inhibition of these metabolizing enzymes causes drug-drug interactions and consequently can lead to serious adverse effects due to bio-accumulation of the drug or its metabolite [76]. Only hesperetin inhibited all the CYP450 superfamilies (CYP1A2, CYP2C19, CYP2C9, and CYP2D6) except for CYP3A4. The administration of this drug candidate could affect its metabolism and excretion, leading to accumulation. Conversely, ECDG, hydroxysaflor yellow A, α-KGA, gallic acid, phloroglucinol, and oleanolic acid showed excellent metabolic profiles and will be easily excreted from the body.
The toxicity of compounds is a significant and crucial parameter in the process of drug approval. Disappointingly, all reported compounds with the exception of α-KGA, gallic acid, catechin, phloroglucinol, oleanolic acid, ursolic acid, acteoside, and osmanthuside B6 are hepatotoxic. However, it is worthy of note that only ZINC63884379 is carcinogenic, which potentially could cause cancer. The human ether-a-go-go (HERG) is a K+ channel found in the heart muscle and regulates the rhythm of the heart. If a drug inhibits HERG, it could result in cardiac arrhythmia and eventually death [77, 78]. In the ADMET table, α-KGA, gallic acid, catechin, hesperetin, ZINC39371167, phloroglucinol, oleanolic acid, ursolic acid, BDC, Quercetin glycoside, and osmanthuside B6 are the only drug candidates with no inhibition of the HERG; hence, they are less likely to cause cardiac arrhythmia and death. All compounds with the exception of ECDG and quercetin glycoside (II), have good acute oral toxicity values. Table 3 shows the toxicity profile of peptides. Interestingly, our result revealed that all peptides are safe for administration, as none of them was found to be toxic.
It is crystal clear from Table 2 that α-KGA, gallic acid, catechin, oleanolic acid, phloroglucinol, and ursolic acid are the best drug candidates with excellent pharmacokinetic and pharmacodynamic profiles. We also discovered that these α-glucosidase inhibitors depicted better ADMET properties than the clinically approved type 2 anti-diabetic drug (acarbose). Hence, they could represent a better alternative to type 2 anti-diabetic drugs. Among the reported small molecule inhibitors of ACE (Table 1), Acteoside and osmanthuside B6 demonstrated an excellent ADMET profile with similar parameters compared to the standard clinically approved lisinopril drug.
Conclusion
In this review, we summarized recently in silico drug discovery findings on α-glucosidase and ACE inhibitors, which could be used as pharmacological intervention in the treatment/prevention of T2D and diabetic chronic complications (hypertension and diabetic nephropathy), respectively. A total of 57 promising compounds that could potentially inhibit α-glucosidase or ACE have been identified. Most of the examples of α-glucosidase and ACE inhibitors cited are competitive and peptide inhibitors, respectively. Furthermore, our review highlighted that most authors do not consider the pharmacokinetic and pharmacodynamic screening of potential lead compounds, which could be pivotal in determining their safety profile. It is worthy of note that most lead compounds failed to pass through clinical trials because of poor pharmacokinetic and unacceptable toxicological profiles. Hence, we investigated the ADMET profile of 27 reported small molecule inhibitors using in silico meta-analysis. Our results revealed that among the α-glucosidase inhibitors, α-KGA, gallic acid, catechin, oleanolic acid, phloroglucinol, and ursolic acid demonstrated excellent ADMET properties while acteoside and osmanthuside B-6 showed good pharmacokinetic and pharmacodynamic properties as compounds with ACE inhibitory prowess. In addition, we evaluated the toxicity profile of the reported peptides and interestingly, all proved to be non-toxic. Moreover, after comparing both sets of inhibitors with clinically approved drugs (acarbose and lisinopril), we found them to have better or similar ADMET profiles, suggesting that they could be used as a better alternative in the management of T2D and its complications. Finally, we propose that further studies be conducted on these potential lead compounds to demonstrate their clinical relevance.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Adelusi Temitope Isaac, Oyedele Abdul-Quddus Kehinde; Writing: Oyedele Abdul-Quddus Kehinde, Boyenle Ibrahim Damilare, Adelusi Temitope Isaac, Abdeen Tunde Ogunlana; Review: Boyenle Ibrahim Damilare, Adelusi Temitope Isaac. Editing: Ashiru Mojeed Ayoola, Atanda Opeyemi Emmanuel, Oyedele Abdul-Quddus Kehinde.
Conflict of interest
The authors declared no conflict of interest.
References
- Baynes HW. Classification, Pathophysiology, Diagnosis and Management of Diabetes Mellitus. J Diabetes Metab. 2015; 6(5):541. [DOI:10.4172/2155-6156.1000541]
- Vinoth Kumar T, Lakshmanasenthil S, Geetharamani D, Marudhupandi T, Suja G, Suganya P. Fucoidan--a α-d-glucosidase inhibitor from sargassum wightii with relevance to type 2 diabetes mellitus therapy. Int J Biol Macromol. 2015; 72:1044-7. [DOI:10.1016/j.ijbiomac.2014.10.013] [PMID]
- BACHEM Holding AG. 2017. Peptides for diabetes research. Accessed July 27, 2018. http://documents.bachem.com/peptides_and_diabetes. Pdf.
- Gong Y, Qin XY, Zhai YY, Hao H, Lee J, Park YD. Inhibitory effect of hesperetin on α-glucosidase: Molecular dynamics simulation integrating inhibition kinetics. Int J Biol Macromol. 2017; 101:32-9. [DOI:10.1016/j.ijbiomac.2017.03.072] [PMID]
- Rahman N, Muhammad I, Gul-E-Nayab, Khan H, Aschner M, Filosa R, et al. Molecular docking of isolated alkaloids for possible α-glucosidase inhibition. Biomolecules. 2019; 9(10):544. [DOI:10.3390/biom9100544] [PMID] [PMCID]
- Ali Asgar MD. Anti-diabetic potential of phenolic compounds: A review. Int J Food Prop. 2013; 16(1):91-103. [DOI:10.1080/10942912.2011.595864]
- Sheliya MA, Rayhana B, Ali A, Pillai KK, Aeri V, Sharma M, et al. Inhibition of α-glucosidase by new prenylated flavonoids from euphorbia hirta L. herb. J Ethnopharmacol. 2015; 176:1-8. [DOI:10.1016/j.jep.2015.10.018] [PMID]
- Kim JH, Kim HY, Yang SY, Kim JB, Jin CH, Kim YH. Inhibitory activity of (-)-epicatechin-3,5-O-digallate on α-glucosidase and in silico analysis. Int J Biol Macromol. 2018; 107(PtA):1162-7. [DOI:10.1016/j.ijbiomac.2017.09.091] [PMID]
- Salehi B, Ata A, V Anil Kumar N, Sharopov F, Ramírez-Alarcón K, Ruiz-Ortega A, et al. Antidiabetic potential of medicinal plants and their active components. Biomolecules. 2019; 9(10):551. [DOI:10.3390/biom9100551] [PMID] [PMCID]
- Kidney Disease Outcomes Quality Initiative (KDOQI). KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007; 49(2 Suppl 2):S12-154. [DOI:10.1053/j.ajkd.2006.12.005] [PMID]
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med. 1999; 341(15):1127-33. [DOI:10.1056/NEJM199910073411506] [PMID]
- Cooper ME. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet. 1998; 352(9123):213-9. [DOI:10.1016/S0140-6736(98)01346-4] [PMID]
- Wolf G, Mueller E, Stahl RA, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest. 1993; 92(3):1366-72. [DOI:10.1172/JCI116710] [PMID] [PMCID]
- Kalinyak JE, Sechi LA, Griffin CA, Don BR, Tavangar K, Kraemer FB, et al. Teh renin-angiotensin system in streptozotocin-induced diabetes mellitus in teh rat. J Am Soc Nephrol. 1993; 4:1337-45. [DOI:10.1681/ASN.V461337] [PMID]
- Lewis E, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851-60. [DOI:10.1056/NEJMoa011303] [PMID]
- Wu HY, Huang JW, Lin HJ, Liao WC, Peng YS, Hung KY, et al. Comparative effectiveness of renin-angiotensin system blockers and other antihypertensive drugs in patients with diabetes: Systematic review and bayesian network meta-analysis. BMJ. 2013; 347:f6008. [DOI:10.1136/bmj.f6008] [PMID] [PMCID]
- Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003; 46(1):3-19. [DOI:10.1007/s00125-002-1009-0] [PMID]
- Hur SJ, Lim BO, Decker EA, McClements DJ. In vitro human digestion models for food applications. Food Chem. 2011; 125(1):1-2. [DOI:10.1016/j.foodchem.2010.08.036]
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American diabetes association and the European association for the study of diabetes. Diabetes Care. 2009; 32(1):193-203. [DOI:10.2337/dc08-9025] [PMID] [PMCID]
- Oboh G, Isaac AT, Akinyemi AJ, Ajani RA. Inhibition of key enzymes linked to type 2 diabetes and sodium nitropusside induced lipid peroxidation in rats’ pancreas by phenolic phenolic extracts of avocardo pear leaves and fruit. Int J Biomed Sci. 2014; 10(3):208-16. [PMID] [PMCID]
- Martinez G, Al-Dalain SM, Menendez S, Guiliani A, Leon OS. Ozone treatment reduces blood oxidative stress and pancreas damage in a streptozotocin-induced diabetes model in rats. Acta Farm Bonaerense. 2005; 24(4):491-7. [Link]
- Atkin M, Laight D, Cummings MH. The effects of garlic extract upon endothelial function, vascular inflammation, oxidative stress and insulin resistance in adults with type 2 diabetes at high cardiovascular risk. A pilot double blind randomized placebo controlled trial. J Diabetes Complications. 2016; 30(4):723-7. [DOI:10.1016/j.jdiacomp.2016.01.003] [PMID]
- Sowers JR, Lester MA. Diabetes and cardiovascular disease. Diabetes Care. 1999; 22 Suppl 3:C14-20. [PMID]
- Ortiz-Andrade RR, García-Jiménez S, Castillo-España P, Ramírez-Avila G, Villalobos-Molina R, Estrada-Soto S. Alpha-glucosidase inhibitory activity of the methanolic extract from tournefortia hartwegiana: An anti-hyperglycemic agent. J Ethnopharmacol. 2007; 109(1):48-53. [DOI:10.1016/j.jep.2006.07.002] [PMID]
- Jandeleit-Dahm KA, Tikellis C, Reid CM, Johnston CI, Cooper ME. Why blockade of the renin-angiotensin system reduces the incidence of new-onset diabetes. J Hypertens. 2005; 23(3):463-73. [DOI:10.1097/01.hjh.0000160198.05416.72] [PMID]
- Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab. 2004; 30(6):487-96. [DOI:10.1016/S1262-3636(07)70146-5] [PMID]
- Cole BK, Keller SR, Wu R, Carter JD, Nadler JL, Nunemaker CS. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension. 2010; 55(3):715-21. [DOI:10.1161/HYPERTENSIONAHA.109.148049] [PMID] [PMCID]
- Furuhashi M, Ura N, Takizawa H, Yoshida D, Moniwa N, Murakami H, et al. Blockade of the renin-angiotensin system decreases adipocyte size with improvement in insulin sensitivity. J Hypertens. 2004; 22(10):1977-82. [DOI:10.1097/00004872-200410000-00021] [PMID]
- Henriksen EJ, Jacob S, Kinnick TR, Teachey MK, Krekler M. Selective angiotensin II receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension. 2001; 38(4):884-90. [DOI:10.1161/hy1101.092970] [PMID]
- Tikellis C, Wookey PJ, Candido R, Andrikopoulos S, Thomas MC, Cooper ME. Improved islet morphology after blockade of the renin-angiotensin system in the ZDF rat. Diabetes. 2004; 53(4):989-97. [DOI:10.2337/diabetes.53.4.989] [PMID]
- Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet. 2002; 359(9311):995-1003. [PMID] [DOI:10.1016/S0140-6736(02)08089-3]
- Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, et al. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The captopril prevention project (CAPPP) randomised trial. Lancet. 1999; 353(9153):611-6. [DOI:10.1016/S0140-6736(98)05012-0]
- Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: The value randomised trial. Lancet. 2004; 363(9426):2022-31. [DOI:10.1016/S0140-6736(04)16451-9] [PMID]
- Nagib AM, Elsayed Matter Y, Gheith OA, Refaie AF, Othman NF, Al-Otaibi T. Diabetic nephropathy following posttransplant diabetes mellitus. Exp Clin Transplant. 2019; 17(2):138-46. [DOI:10.6002/ect.2018.0157] [PMID]
- Cao Z, Cooper ME. Pathogenesis of diabetic nephropathy. J Diabetes Investig. 2011; 2(4):243-7. [DOI:10.1111/j.2040-1124.2011.00131.x] [PMID] [PMCID]
- Vinod PB. Pathophysiology of diabetic nephropathy. Clin Queries Nephrol. 2012; 1(2):121-6. [DOI:10.1016/S2211-9477(12)70005-5]
- Sowers JR, Epstein M. Diabetes mellitus and associated hypertension, vascular disease, and nephropathy: An update. Hypertension. 1995, 26:869-79. [DOI:10.1161/01.HYP.26.6.869] [PMID]
- Strippoli GF, Craig M, Craig JC. Antihypertensive agents for preventing diabetic kidney disease. Cochrane Database Syst Rev. 2005; (4):CD004136. [DOI: 10.1002/14651858.CD004136.pub2] [PMID]
- Ren L, Qin X, Cao X, Wang L, Bai F, Bai G, et al. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell. 2011; 2(10):827-36. [DOI:10.1007/s13238-011-1105-3] [PMID] [PMCID]
- Sim L, Quezada-Calvillo R, Sterchi EE, Nichols BL, Rose DR. Human intestinal maltase-glucoamylase: Crystal structure of the n-terminal catalytic subunit and basis of inhibition and substrate specificity. J Mol Biol. 2008; 375(3):782-92. [DOI:10.1016/j.jmb.2007.10.069] [PMID]
- Xu Y, Lee J, Park YD, Yang JM, Zheng J, Zhang Q. Molecular dynamics simulation integrating the inhibition kinetics of hydroxysafflor yellow a on α-glucosidase. J Biomol Struct Dyn. 2018; 36(4):830-40. [DOI:10.1080/07391102.2017.1300544] [PMID]
- Xiong SL, Lim GT, Yin SJ, Lee J, Lee JR, Hahn MJ, et al. Inhibitory effect of α-ketoglutaric acid on α-glucosidase: Integrating molecular dynamics simulation and inhibition kinetics. J Biomol Struct Dyn. 2020; 38(12):3496-503. [DOI:10.1080/07391102.2019.1659858] [PMID]
- Choudhary DK, Chaturvedi N, Singh A, Mishra A. Characterization, inhibitory activity and mechanism of polyphenols from faba bean (gallic-acid and catechin) on α-glucosidase: Insights from molecular docking and simulation study. Prep Biochem Biotechnol. 2020; 50(2):123-32. [DOI:10.1080/10826068.2019.1679171] [PMID]
- Choudhary MI, Adhikari A, Rasheed S, Marasini BP, Hussain N, Kaleem WA. Cyclopeptide alkaloids of ziziphus oxyphylla edgw as novel inhibitors of α-glucosidase enzyme and protein glycation. Phytochem Lett. 2011; 4(4):404-6. [DOI:10.1016/j.phytol.2011.08.006]
- Tiong SH, Looi CY, Hazni H, Arya A, Paydar M, Wong WF, et al. Antidiabetic and antioxidant properties of alkaloids from catharanthus roseus (l.) g. don. Molecules. 2013; 18(8):9770-84. [DOI:10.3390/molecules18089770] [PMID] [PMCID]
- Laoud A, Ferkous F, Maccari L, Maccari G, Saihi Y, Kraim K. Identification of novel nt-MGAM inhibitors for potential treatment of type 2 diabetes: Virtual screening, atom based 3D-QSAR model, docking analysis and ADME study. Comput Biol Chem. 2018; 72:122-35. [DOI:10.1016/j.compbiolchem.2017.12.003] [PMID]
- Zafar M, Khan H, Rauf A, Khan A, Lodhi MA. in silico study of alkaloids as α-glucosidase inhibitors: Hope for the discovery of effective lead compounds. Front Endocrinol. 2016; 7:153. [DOI:10.3389/fendo.2016.00153] [PMID] [PMCID]
- Mollica A, Zengin G, Durdagi S, Ekhteiari Salmas R, Macedonio G, Stefanucci A, et al. Combinatorial peptide library screening for discovery of diverse α-glucosidase inhibitors using molecular dynamics simulations and binary QSAR models. J Biomol Struct Dyn. 2019; 37(3):726-40. [DOI:10.1080/07391102.2018.1439403] [PMID]
- Wan JX, Lim G, Lee J, Sun XB, Gao DY, Si YX, et al. Inhibitory effect of phloroglucinol on α-glucosidase: Kinetics and molecular dynamics simulation integration study. Int J Biol Macromol. 2019; 124:771-9. [DOI:10.1016/j.ijbiomac.2018.11.268] [PMID]
- Ding H, Hu X, Xu X, Zhang G, Gong D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int J Biol Macromol. 2018; 107(Pt B):1844-55. [DOI:10.1016/j.ijbiomac.2017.10.040] [PMID]
- Meena SN, Kumar U, Naik MM, Ghadi SC, Tilve SG. α-Glucosidase inhibition activity and in silico study of 2-(benzo[d][1,3]dioxol-5-yl)-4H-chromen-4-one, a synthetic derivative of flavone. Bioorg Med Chem. 2019; 27(12):2340-4. [DOI:10.1016/j.bmc.2018.12.021] [PMID]
- Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, et al. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci U S A. 1988; 85(24):9386-90. [DOI:10.1073/pnas.85.24.9386] [PMID] [PMCID]
- Ehlers MR, Riordan JF. Angiotensin-converting enzyme: New concepts concerning its biological role. Biochemistry. 1989; 28(13):5311-8. [DOI:10.1021/bi00439a001] [PMID]
- Fuchs S, Xiao HD, Hubert C, Michaud A, Campbell DJ, Adams JW, et al. Angiotensin-converting enzyme c-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension. 2008; 51(2):267-74. [DOI:10.1161/HYPERTENSIONAHA.107.097865] [PMID]
- Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995; 270(8):3656-61. [DOI: 10.1074/jbc.270.8.3656] [PMID]
- Li P, Xiao HD, Xu J, Ong FS, Kwon M, Roman J, et al. Angiotensin-converting enzyme N-terminal inactivation alleviates bleomycin-induced lung injury. Am J Pathol. 2010; 177(3):1113-21. [DOI:10.2353/ajpath.2010.081127] [PMID] [PMCID]
- Yu Z, Fan Y, Zhao W, Ding L, Li J, Liu J. Novel angiotensin-converting enzyme inhibitory peptides derived from oncorhynchus mykiss nebulin: Virtual screening and in silico molecular docking study. J Food Sci. 2018; 83(9):2375-83. [DOI:10.1111/1750-3841.14299] [PMID]
- De Oliveira TV, Guimarães AP, Bressan GC, Maia ER, Coimbra JSDR, Polêto MD, et al. Structural and molecular bases of angiotensin-converting enzyme inhibition by bovine casein-derived peptides: An in silico molecular dynamics approach. J Biomol Struct Dyn. 2021; 39(4):1386-1403. [DOI:10.1080/07391102.2020.1730243] [PMID]
- Tahir RA, Bashir A, Yousaf MN, Ahmed A, Dali Y, Khan S, et al. in silico identification of angiotensin-converting enzyme inhibitory peptides from MRJP1. PloS One. 2020; 15(2):e0228265. [DOI:10.1371/journal.pone.0228265] [PMID] [PMCID]
- Auwal SM, ZainalAbidin N, Zarei M, Tan CP, Saari N. Identification, structure-activity relationship and in silico molecular docking analyses of five novel angiotensin I-converting enzyme (ACE)-inhibitory peptides from stone fish (actinopygalecanora) hydrolysates. PloS One. 2019; 14(5):e0197644. [DOI:10.1371/journal.pone.0197644] [PMID] [PMCID]
- Yu D, Wang C, Song Y, Zhu J, Zhang X. Discovery of novel angiotensin-converting enzyme inhibitory peptides from todarodes pacificus and their inhibitory mechanism: In silico and in vitro studies. Int J Mol Sci. 2019; 20(17):4159. [DOI:10.3390/ijms20174159] [PMID] [PMCID]
- Yu F, Zhang Z, Luo L, Zhu J, Huang F, Yang Z, et al. Identification and molecular docking study of a novel angiotensin-i converting enzyme inhibitory peptide derived from enzymatic hydrolysates of cyclina sinensis. Mar Drugs. 2018; 16(11):411. [DOI:10.3390/md16110411] [PMID] [PMCID]
- Taga Y, Hayashida O, Ashour A, Amen Y, Kusubata M, et al. Characterization of angiotensin-converting enzyme inhibitory activity of X-Hyp-Gly-type tripeptides: Importance of collagen-specific prolyl hydroxylation. J Agric Food Chem. 2018; 66(33):8737-43. [DOI:10.1021/acs.jafc.8b03648] [PMID]
- Ali A, Baby B, Soman SS, Vijayan R. Molecular insights into the interaction of hemorphin and its targets. Sci Rep. 2019; 9(1):14747. [DOI:10.1038/s41598-019-50619-w] [PMID] [PMCID]
- Lin K, Zhang L, Han X, Meng Z, Zhang J, Wu Y, et al. Quantitative structure-activity relationship modeling coupled with molecular docking analysis in screening of angiotensin i-converting enzyme inhibitory peptides from qula casein hydrolysates obtained by two-enzyme combination hydrolysis. J Agric Food Chem. 2018; 66(12):3221-8. [DOI:10.1021/acs.jafc.8b00313] [PMID]
- Chamata Y, Watson KA, Jauregi P. Whey-derived peptides interactions with ACE by molecular docking as a potential predictive tool of natural ACE inhibitors. Int J Mol Sci. 2020; 21(3):864. [DOI:10.3390/ijms21030864] [PMID] [PMCID]
- Muhammad SA, Fatima N. In silico analysis and molecular docking studies of potential angiotensin-converting enzyme inhibitor using quercetin glycosides. Pharmacogn Mag. 2015; 11(Suppl 1):S123-6. [DOI:10.4103/0973-1296.157712] [PMID] [PMCID]
- Arya H, Syed SB, Singh SS, Ampasala DR, Coumar MS. In silico investigations of chemical constituents of clerodendrum colebrookianum in the anti-hypertensive drug targets: ROCK, ACE, and PDE5. Interdiscip Sci. 2018; 10(4):792-804. [DOI:10.1007/s12539-017-0243-6] [PMID]
- Honório KM, Moda TL, Andricopulo AD. Pharmacokinetic properties and in silico ADME modeling in drug discovery. Med Chem. 2013; 9(2):163-76. [DOI:10.2174/1573406411309020002] [PMID]
- Kubinyi H. Drug research: Myths, hype and reality. Nat Rev Drug Discov. 2003; 2(8):665-8. [DOI:10.1038/nrd1156] [PMID]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004; 3(8):711-5. [DOI:10.1038/nrd1470] [PMID]
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, et al. Pubchem substance and compound databases. Nucleic Acids Res. 2016; 44(D1):D1202-13. [DOI:10.1093/nar/gkv951] [PMID] [PMCID]
- Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, et al. Admetsar 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019; 35(6):1067-9. [DOI:10.1093/bioinformatics/bty707] [PMID]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source Drug Discovery Consortium, et al. In silico approach for predicting toxicity of peptides and proteins. PloS One. 2013; 8(9):e73957. [DOI:10.1371/journal.pone.0073957] [PMID] [PMCID]
- Jayaraj JM, Reteti E, Kesavan C, Muthusamy K. Structural insights on vitamin d receptor and screening of new potent agonist molecules: Structure and ligand-based approach. J Biomol Struct Dyn. 2021; 39(11):4148-59. [DOI: 10.1080/07391102.2020.1775122] [PMID]
- Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, et al. New era in drug interaction evaluation: US food and drug administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008; 48(6):662-70. [DOI:10.1177/0091270007312153] [PMID]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995; 81(2):299-307. [DOI:10.1016/0092-8674(95)90340-2] [PMID]
- Aronov AM. Predictive in silico modeling for hERG channel blockers. Drug Discov Today. 2005; 10(2):149-55. [DOI:10.1016/S1359-6446(04)03278-7]
Type of Study: Review article |
Subject:
Mollecular Modeling
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information